Absorption spectroscopy refers to spectroscopic techniques that measure the absorption of radiation, as a function of frequency or wavelength, due to its interaction with a sample. The technique uses basically the principle that free atoms (gas) generated in an atomizer can absorb radiation at a specific frequency. The sample absorbs energy, i.e., photons, from the radiating field. It is a molecular spectroscopy method that uses the wavelength-dependent absorption characteristics of materials to identify and quantify specific substances. The intensity of the absorption varies as a function of frequency, and this variation is the absorption spectrum. The absorbance of a solution increases as the attenuation of the optical beam increases.
Absorption spectroscopy is employed as an analytical chemistry tool to determine the presence of a particular substance in a sample and, in many cases, to quantify the amount of the substance present. Infrared and ultraviolet-visible spectroscopy is particularly common in analytical applications. It is also employed in studies of molecular and atomic physics, astronomical spectroscopy and remote sensing.
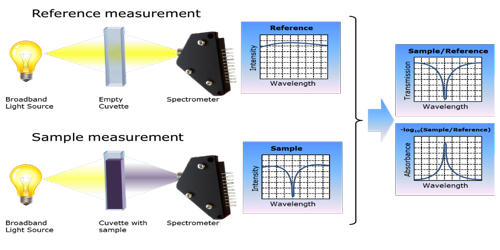
The principle of absorption spectroscopy is to measure how much light is absorbed by the sample. There is a wide range of experimental approaches for measuring absorption spectra. The most common arrangement is to direct a generated beam of radiation at a sample and detect the intensity of the radiation that passes through it. It measures the loss of electromagnetic energy after it illuminates the sample under study. The transmitted energy can be used to calculate the absorption. Atomic-absorption spectroscopy quantifies the absorption of ground-state atoms in the gaseous state.
Applications
There are many different approaches for measuring absorption spectra. Absorption spectroscopy is useful in chemical analysis because of its specificity and its quantitative nature. The most common one is to point a generated beam of light at a sample and detect the intensity of the radiation that goes through it. The specificity of absorption spectra allows compounds to be distinguished from one another in a mixture, making absorption spectroscopy useful in a wide variety of applications. The energy that is then transmitted is used to calculate the absorption. For instance, Infrared gas analyzers can be used to identify the presence of pollutants in the air, distinguishing the pollutant from nitrogen, oxygen, water, and other expected constituents.
Absorption spectroscopy relies on the absorption of light by biomolecules at a particular wavelength. The specificity also allows unknown samples to be identified by comparing a measured spectrum with a library of reference spectra. In many cases, it is possible to determine qualitative information about a sample even if it is not in a library. In this process, the energy of a photon is taken up by the electrons of the atoms in matter. Infrared spectra, for instance, have characteristics absorption bands that indicate if carbon-hydrogen or carbon-oxygen bonds are present. It can be used with any type of wave, including infrared, gamma, microwave, x-ray, visible light, sound, atomic, even radio waves.