A review on solid dispersion:
A substitute approach for poorly water soluble drug
An amount of methodologies can be adapted to progress solubility and bioavailability of poorly soluble drugs. The procedure is worn for the solubilization of drug includes solid dispersion, micronization, chemical modification, PH adjustment, complexation,cosolvency, miceller solubilisation, hydrotrophy etc. The idiom solubility is defined as greatest quantity of solute dissolved in the solvent. It can be two types. 1. Qualitative or 2.Quantitative.(Sato et al,1981)Absorption of drugs from gastrointestinal tract is limited by various factors. The most noteworthy contributors being poor aqueous solubility and poor membrane permeability of the drug molecule. When the drug is delivered orally, it must first dissolve in the gastric/intestinal fluid. Consequently the drug with the poor solubility will typically show dissolution rate limited absorption. Solid dispersion technology is used to improve the characteristics of poorly water soluble drugs and oral bioavailability. Different solid dispersion system has been established in the pharmaceutical journalism to progress the dissolution properties of poorly water soluble drugs. It has been anticipated that 40% of new chemical entities presently being revealed are poorly water soluble. (Sekigushi and Obi,1961)) Various strategies have been developed to overcome these problems. These are prodrug formation, complexation, microcapsulation, the use of surfactants, lipids, permeation enhancers, micronization, saltformation, cyclodextrins, nanoparticles, solid dispersion and self emulsifying drug delivery system.(Okamoto and Oguni,1996)The therapeutic response of a drug normally dependent on the adequate concentration of the drug that are achieved and maintained at the site or site of action of the drug. The majority of the drug that is administered orally is intended to be absorbed from the gastrointestinal tract. The rate limiting step may vary from drug to drug (Jain et al,2006). The enhancement of oral bioavailability of these poorly water soluble drugs remains one of the most challenging aspects in the drug development. The biopharmaceutical classification system divides into four classes that is depending on the in vivo permeability data and the in vitro solubility.
Among these classes number II class shows the poor solubility and high permeability. Class II drugs low the ability to dissolve and it is the more important limitation to their overall rate and extent the absorption.(Simonelli andMehta,1969) Then their ability to permeate through the membrane. class II enhance the aqueous solubility or dissolution rate. There are various strategies to improve the dissolution rate and the aqueous solubility. These are solid dispersion, particle size reduction, salt formation, complexation with cyclodextrin, The use of co solvents and self emulsifying agents. Among these all techniques solid dispersion has proven to be very successful.The dissolution rate of the poorly water soluble drug in a solid dispersion is increased by i)increasing the surface area and ii)improving wettability hence decreasing the thickness of the diffusion layer iii)enhancing the solubility of the drug by the formation of the supersaturated solution.(Amidon et al,1995)
Mechanism of solubilization:
Solubility is defined as the maximum concentration of the drug solute that are dissolved in the solvent under the specified temperature,PH, and pressure.the drug solubility is related to the rate of bioavailability.
The solubility of a weak acid or weak base varies with the fraction of PH.
Solubility of weak acid,total solubility(Cs)is given byCs=[HA]+[A-]
Where [HA]=intrinsic solubility of non ionized acid
[A-]=the concentration of its anion.
The anion concentration can be expressed in terms of dissociation constant Ka
Cs=Co+Ka[Co/h=]
Where Co=non ionixed acid concentration
This indicates that the solubility of the weak acid increases with increase in the ph solubility is optimal at higher Ph.
Solubility of a weak base is given by the expression: Cs=Co+[H+/Ka]
It indicates that the solubility of the weak base decreases with increasing PH and the solubility is optimal at the lower ph. (Ford,1986)
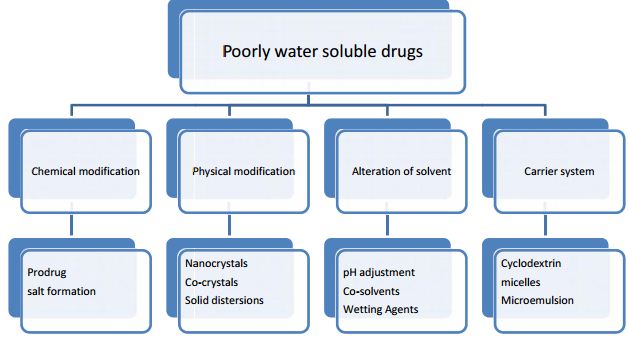
Various approaches for solubility enhancement:
- Drug dispersion in carries:
-Eutectic mixtures
-Solid solutions
-Solid dispersion
- Solubilization with solvent
- Complexation with polymer
- Micronization
- Alteration in PH
- Use of surfactant
- Use of cyclodextrin
- Change in physical form
- Use of prodrug and drug derivatization
Definition of Solid dispersion:
Chiou and Riegelman defined solid dispersion as “a dispersion involving the formation of eutectic mixture of drugs with water soluble carries by melting of their physical mixtures”(Chiou and riegleman,1986).The term solid dispersion refers to the dispersion of one or more active ingredient in an inert carrier or matrix at solid solid state prepared by melting(fusion),solvent, or the melting solvent method. Once the solid dispersion was exposed to aqueous media and the carrier dissolved, the drug is released as very fine, colloidal particles. the term solid dispersion refers to the group of solid products consisting of at least two different components, generally a hydrophobic drug and hydrophilic matrix.(Lidenberg,2004) The matrix can be either crystalline or amorphous. The dispersion of a drug or drugs in a solid diluents or diluents by traditional mechanical mixing is not included in this category. Therefore, based on their molecular arrangements, different types of solid dispersions can be distinguished and combination can be encountered in the same sample i.e some molecules are present in clusters while some are molecularly dispersed. In various studies the type of solid dispersion is based on the method of preparation. So, the solid dispersion refers to the group of solid products consisting of at least two different components generally a hydrophilic matrix and a hydrophobic drug.(Yal et al,1985)
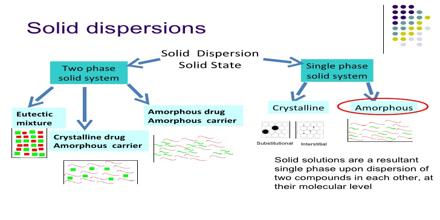
Classification of solid dispersion:
- Simple eutectic mixture
- Solid solution
- Glass solution
- Complex formation
- Amorphous precipitation in a crystalline carrier
Solid solutions:
It is consist of a solid solute dissolved in a solid solvent.The particle size is reduced to molecular level.it was reported that the solid solution of a poorly soluble drug in a fast dissolving carrier achieves a faster dissolution rate than a eutectic mixture because the drug particle size is reduced to its absolute minimum as it is molecularly dispersed in the carrier in a solid solution.The effective surface area is significantly higher hence the dissolution rate is increased.Solid solutions classified by their miscibility characteristics.(Mallet et al,1965)
(1) Continuous solid solutions:
In a continuous solid solutions the components are totally miscible with one another in all proportions in both solid and the liquid state. The lattice energy of the continuous solid solution at all compositions is higher than that of the respective pure components in the solid state, because the heteromolecular bonding is higher than the homomolecular.(Boral,1995)
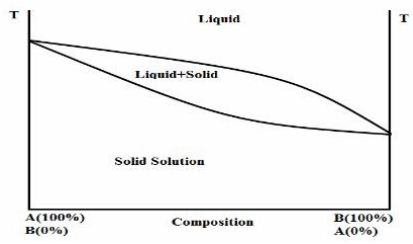
Hypothetical phase diagram of a continuous solid solution (Jain and shakrma,2006)
(2) Discontinuous solid solution:
In discontinuous solid solution the miscibility or solubility of one component in the other is limited. Formulation of dosage form with solid solution will depend on both the mutual solubilities of the two components and dose of the drug component. The maximum limit of a tablet or capsule is about 1 gram. Assuming that the solubility of the drug in the carrier is 10%,doses of above 100 mg would not be feasible with its strategy. If the drug solubility in the carrier is significantly higher than 10%,larger doses can be entertained.(Jain and Sharma 2006)
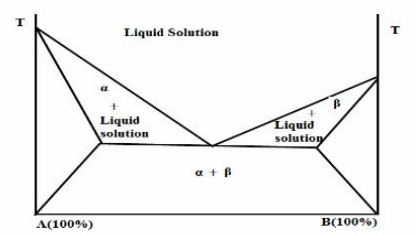
Hypothetical phase diagram of a discontinuous solid solution (Bulter and Mattewk,1999)
- Substitutional solid solution:
Here the solid molecules replace the solvent molecule in the crystal lattice of the solid solvent. An extensive solid solution can only be formed when the effective diameter of the solute differs by less than 15% from that of the solvent and when the packing patterns of solvent and solute are comparable.
Glass solutions:
It is also known as an amorphous solution,is a homogenous system in which a glassy or a vitreous form of carrier solubilities drug molecules. The glassy or vitreous state is characterized by transparency and brittleness below the glass transition temperature. The temperature at which the glassy polymer becomes rubbery on heating and a rubbery polymer reverts to a glassy one on cooling is called the glass transition temperature, Tg.(Mullins and Macek,1960) Well below the Tg, the glass solutions are hard, stiff glassy materials; at the temperature well above Tg, the materials are rubbery. They are formed by either cooling of the melt or evaporation of a solution. Upon cooling or evaporation ,the drug is vitrified into its glassy state in the amorphous carrier. Specific volume, specific heat, viscosity, refractive index, compressibility, thermal conductivity and other physico chemical properties of the glass show changes when cooled or heated through the glass transition temperature.
Complex formation:
Here the drug forms a complex with an inert water soluble carrier in the solid state. The availability of the drug depends on the stability constant of the complex and the absorption rate of the drug. It also depends on the solubility of the drug.Herecyclo dextrins are mostly used. This is linked with the oligosaccharides of alpha –D-glucopyranose containing a relatively hydrophobic central cavity and hydrophilic outer surface. The drugs that are poorly water soluble, Their solubility can be increased by this process.
Amorphous precipitation in a crystalline carrier:
The drug also may precipitate in the amorphous form in the crystalline carrier.Here the energy state is high and this high energy state of the drug generally produces the greater dissolution rate than the corresponding crystalline forms of the drug.(Gines et al,1995)
Advantages of solid dispersion:
By using solid dispersion ,management of the drug release profile is achieved by manipulation of the carrier and solid dispersion particles properties. The parameters are-drug crystallinity, particle porosity, molecular weight and composition and wettability, when it is controlled successfully the bioavailability is improved.
Particles with reduced particle size:
Particle sizes of the particles are reduced during the preparation of the solid dispersion and the surface area is improved. As a result it increases the dissolution rate. However bioavailability is improved.
Particles with improved wettability:
During the preparation of solid dispersion, the wettability is improved. Many carriers are used for the solid dispersion to enhance the wetting properties. So improved wetting may lead to reduced agglomeration and increased surface area.
Particles with higher porosity
Highly degree of porosity have been found in the particles of solid dispersion.The increase in porosity depend on the carrier properties.solid dispersion that contains linear polymer result in a higher dissolution rate. This increased in porosity hastens the drug release profile.
Drugs in amorphous state:
Drugs in amorphous state shows higher release of drug because there is no any energy to break the crystal lattice during the dissolution process.In solid dispersion,drugs are presented as supersaturated solutions.It forms the higher solubility if it is in the metastable polymorphic form.(Butler and Mattew,1999)
Disadvantages of solid dispersion:
- Solid dispersion are not broadly used in the commercial products because duringprocessing or storage there is the possibility to undergo the crystallization from the amorphous state.
- Moisture has the great effect on the storage stability of the amorphous pharmaceuticals because it may increase drug mobility and promote drug crystallization.
- Polymers that are used in the solid dispersion can absorb moisture.And this result in the phase separation,crystal growth or conversion from the amorphous to the crystalline state.This may result in decreased solubility and dissolution rate.
- For the purpose of manufacturing there is poor scale up.
- Expensive method.
- Difficult to incorporate into formulation of dosage forms.
- Scale-up of manufacturing process.
- Stability of the drug and vechicle.
Pharmaceutical applications of solid dispersion:
- To increase the solubility of the poorly soluble drugs that increases the dissolution rate, bioavailability and the absorption.
- To obtain the homogeneous distribution of drugs in solid state.
- To protect against the decomposition and stabilize the unstable drugs by different process such as oxidation, hydrolysis, racemization, photo oxidation etc.
- To dispense liquid or gaseous compounds.
- To reduce the side effects of drugs.
- To formulate the sustained release preparation of soluble drugs by dispersing the poorly soluble or insoluble carrier.
- To formulate the fast release priming drug.
- To mask the unpleasant smell, odor.
- To avoid undesirable incompatibilities.
- To reduce pre systemic inactivation of drugs like morphine.
MATERIALS AND METHODS
Preparation of solid dispersion:
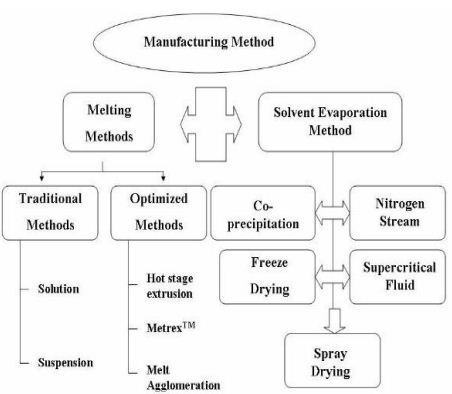
Many techniques are now used for the preparation of the solid dispersion.But phase separation and demixing are the most common problem in the preparation of solid dispersion.It was reported that that the cooling process in the rapid way reduce the extent of phase separation .The techniques are-
Solvent evaporation method:
In this method there are two important steps:
The solution containing the matrix and the drug is prepared by proper molecular level mixing with the solvent.
Removal of the solvent:
The aim of this method is to dissolve the drug and the carrier.This should be done simultaneously in a common solvent,this is characterized by the removal of solvent by evaporation.Thetemperature is usually-23-65oc.The solvent also can be removed by the freeze drying or spray drying.The drug that are used in the solid dispersion are normally hydrophobic and the carrier is hydrophilic.Sometimes it becomes difficult to identify a common solvent to dissolve both components.(Martin et al,2003)
Hot melt extrusion method:
This method was introduced in 1970s as the manufacturing tool in the pharmaceutical industry.The basic principle of this technology is similar to the fusion method except for the employing of extruder for high shear mixing.This method is very common in the processing plastics in the polymer industry.This has been useful in the preparation of solid dispersion in the single step.(Sekiguchi and Obi,1961) It is also very important for the preparation of various dosage forms in the pharmaceutical industry such as preparation of sustained release pellets.To form solid dispersion of various drugs during hot melt extrusion,polymers such as PVP,HPMC acetate succinate etc were used.
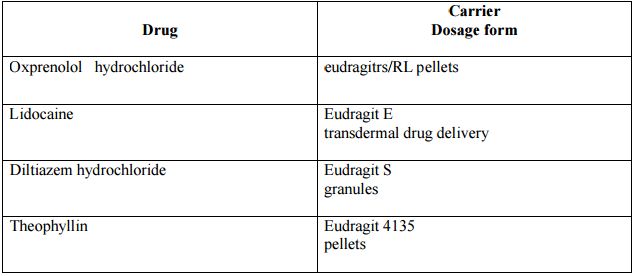
List of drugs and polymers used during the preparation:
Supercritical fluid technology:
This technology was introduced in the late 1980s and early 1990s.It is any substance at a temperature and pressure above its critical point.It can diffuse through the solids like a gas and like a liquid dissolve the materials.If the critical point is close to it,there is small changes in the temperature and the pressure and this result large changes in density.(Mura et al,1996)
Supercritical fluid technology is suitable as a substitute for organic solvents in a range of laboratory processes and industry. Depending on the method different applications have been introduced. These are:
- Precipitation from supercritical solutions-rapid expansion of supercritical solution(RESS)
- Gas antisolvent or supercritical antisolvent(SAS) recrystallization
- Solution enhanced dispersion by supercritical fluids(SEDS)
- Aerosol solvent interaction system
- Precipitation with compressed antisolvent(OCA)
- Presipitation from gas saturated solutions(PGSS)
Direct capsule filling:
Francois and Jones was first described this method.They described that filling of semisolid materials into hard gelatin capsules as melts,which occur in the room temperature.For this reason Chathan prepared PEG-based solid dispersion by filling the PEG melt in the hard gelatin capsule.Then the molten dispersion was cooled to the room temperature.But PEG is not the suitable carrier for the direct capsule filling because the water soluble carrier dissolved more rapidly than the drug.As a result rich layer was formed on the surface which prevent further dissolution of drug.(Sharma and Joshi,2007)
Use of surfactants:
Use if surfactant for solid dispersion has been increased in these recent years.Serajuddin achieved a complete dissolution of drug from solid dispersion by using surface active or surface emulsifying carriers.These carriers prevent the formation of water insoluble surface layers by dispersing which resulted in a high surface of the drug.This facilitate the gastrointestinal fluids in the presence of bile salt,lecithin.
The commonly used surface active agents are-gelucire44/14,vitamin E TPGS NF and Tween 80.(Zerrouk et al,2001)
Electrostatic spinning:
In this process a drug matrix solution is pumped through an orifice and that is then used for the electrical field.when electrical forces overcome the surface tension of the drug then fibers of submicron diameters are formed.When the solvents evaporate then the fibers are collected.Thiselectrospunfibres are incorporated into hard gelatin capsule.The diameter of the fiber can be adjusted by surface tension,electrical field and dielectric constant.After rapid evaporation of the fiber,these fibers are used for the further process.(Wang et al,2005)
Melt agglomeration:
A processor has been used for the preparation of solid dispersion by this melt agglomeration method and this processor is rotary processor.In this process binder acts as the carrier.Solid dispersion are prepared by either heating binder,drug and excipient to a temperature above the melting point of the binder.Molten binder are heated excipient by using high shear mixing.(okonogi,2006) It has been reported that the melt in procedure gives a higher dissolution rate.Melt in procedure also results in homogenous distribution of drug in agglomerate.(Nokhodchi,2007)
Cryogenic technique:
It is reported that all the fuids have a critical temperature which lies below or within the normal ambient temperature range and this is defined as cryogenic.The upper limit of the normal ambient range is 323.18 degree Kelvin. Based on this concept, cryogen are gases that can not exist as liquid, without refrigeration. In the family of cryogens it begins with Hillium II near the lower boundary and carbon di oxide is included. This techniques has developed to enhance the dissolution rate by creating nonstructured amorphous particles with high degree of porosity. There are various cryogenic technique for the preparation of solid dispersion particles. These are
- Spray freezing onto cryogenic fluids
- Spray freezing into cryogenic fluids
- Spray freeze drying
- Ultra rapid freezing
Characterization of solid dispersion:
Thermal analysis:
This is used for the study of the physicochemical interaction of two or more component system.
Cooling curve method:
Until a homogenous melt are obtained the physical mixtures of various compositions are heated. The temperature is recorded. There are many disadvantages in this method .it is time consuming, it requires large amount of solvent, changes in slopes can be missed. it occurs if the cooling is done very rapidly. Another is this method can not be applied to samples that decompose after melting. This is very difficult to detect the samples when there is small solid solid solubility. This method is recently used for the determination of phase diagrams of deoxycholic acid menadione.(Guillory et al,1969)
Thaw-melt method:
A capillary melting tube is taken and that is filled with a sample of solidified mixture. Then it is heated gradually. Thaw point is referred to the temperature on crossing a solidus line. A stirring device in a capillary tube gives the more accurate results.(Moore,1969) This stirring facilitates the homogenous system and this only affect on the melting point but not in the thaw point. In the comparison of the simple eutectic system and the limited solid solution, there is a diagnostic line in the thaw point. There is no need to use complicated device.(Sekiguchi and Obi,1964)
Thermo microscopic method:
To study the phase diagram of binary system ,a hot stage of polarized microscopy was used.The physical mixture is placed on the slide cover with a cover slip this is sealed .For the sealing silicone grease is used which prevent the sublimation. Until the mixture is liquefies, the mixture is heated. After cooling the mixture is again heated for 4 mins. By the visual observation then the thaw and the melting point are then determined. The main advantage of this method is, it is simple and it requires less sample.(R.E, 1964) But there are some disadvantages too.It suffers from being subjective, limited to thermally stable compounds and potentially inhomogeneous in distribution after resolidification.
DTA:
For studying the phase equilibria of either a pure compound or a mixture DTA is an effective thermal method .it is associated with physical or chemical changes. The temperature or time is recorded automatically as the substance is heated at a uniform time. Polymorphic, transitions, evaporations, sublimation desolvation and other types of decomposition can be detected by this DTA (Wendlandt,1964). The main advantage of this method is in constructing phase diagrams of high reproducibility. Although there are some sensitivity and accuracy of the DTA like sample size, heatingrate, sample geometry, thermal conductivity etc but these variables can be optimize the desired characteristics of the DTA apparatus. This is especially a valuable method for detecting the presence of small amount of eutectic in the mixture (Sekiguchi et al,1964)
Zone melting method:
In 1952 this technique was first introduced (Pfann,1966). It is primarily used for the ultrapurification of metals and inorganic and organic compounds. For metal and organic and inorganic compounds the phase diagram can be detected. For the mixing of the liquid in the melting zone a mechanical stirring device is also required.The bar is sectioned and analyzed for its chemical composition, after the zone melting is finished. Aphase diagram of binary or multicomponent system can be constructed from the temperature and chemical composition.
This method is limited to the compounds that have high thermal solubility and low volatility (Pfann,1966).It is important for the determination of exact chemical composition of a eutectic and the minute solid- solid solubility.By this method many phase diagrams has been detected.
X-ray diffraction method:
This method is used to measure the intensity of the Xray diffraction from different angle. There are both the advantages and the disadvantages of this method. A better resolution of diffraction peaks can be obtained in the former method.It is easy to determine the relative diffraction intensity.on the other side,it requires more sample and less realibility (Wald and Rosenberg,1965) It is also more sensitive to sample preparation and position. For studying the physical nature of solid dispersion this method is more important. It is also used for the study of binary eutectic tool of chloramphenicol-urea griseofulvin succinic acid. Many phase diagrams of inorganic and metal compounds can be detected by this method. Diffraction peaks of each crystalline components can be found in diffraction spectra are obtained from the simple eutectic system.(Mallet and Mullet,1965)
Microscopic method:
Microscopic method is used in the study of polymorphism and in the morphology of solid dispersion. By the polarizing microscope ,the fine particles of crystallization in the glass polyvinyl pyrrolidone matrix can be detected. For this process a high resolution of an electron microscope was used. The limitation of this technique is ,it is not suitable for the chemicals with high atomic numbers.
Spectroscopic method:
Visible absorption spectroscopy is used to measure the low concentration of beta carotene.It is used in polyvinyl pyrrolidone.It is dissolved in organic solvents but the beta particles are not dissolved. This indicates that beta carotene is dispersed molecularly in the polymer.IR spectroscopy is also used in the study of solid solution of nitrile ion in many inorganic compounds such as KBR,nacl etc.(Moore,1964)
Dissolution rate method:
It is the recent method which is proposed by Allen and Kwan.This method is developed to study the degree of crystallinity which is mainly in solid-solid equilibria,especially in temperature regions below solid-liquid equilibria.The methods involve in vitro method (Kakumanu and Bansal,2003) .The technique is simple to perform,except some binary system. In the binary system the tablet surface may not be remain constant.
Thermodynamic method:
By this parameter the phase diagram of eutectic and solid solutions system can be constructed.Also the solubility gap of agbr-nabr system was found from the thermodynamic data that is obtained from the electromotive force study by galvanic cell.(Rastogi,1956)
Aging of solid dispersion:
The solid dispersion act as the potential dosage form of medication for increasing dissolution and absorption rates of poorly soluble drugs.However this results the aging or storage under various conditions and it effects the fast release characteristics and chemical stabilities, that have not been reported (Ahmed,1993).The interesting and important research of this method is feasible. There are many effects on non pharmaceuticals systems also. This method has effect on dissolution andbropirimine whereas such ageing has no effect on the dissolution of drug.(Chiou and Riegelman,1971)
Aging effects of eutectic mixture:
As we know that the dispersed phase particles tend to the coarsen on aging.This is because the interfacial energy of the system is reduced by the concomitant reduction in interface area. This system is occurred in eutectic system with or without solid solution formation. The extent increases with the time and the aging temperature.(Rastogi and Bassi,1964)
Aging effect on glass solution:
As we know that glass solution is a metastable form and it is subjected to the aging transformation, forming a more stable form.This process is occur more rapidly or extremely slowly because of the untreated ordinary window glass kept at the room temperature.Small angle Xray scattering and electrone microscope methods were used to study the kinetics. (Mallet and Fay,1965)
Aging effects of solid solution:
There is the most important aging effect from the solid solutions is the precipitation from the solid solutions that is supersaturated along with the physical and the chemical properties. There are some hardening effect and those are time,temperature and the function of composition. The preparation of aging for the long time at a given temperature may also cause them to lose their hardness. (Smoluchowski,1951)
Suitable properties of a carrier for solid dispersions:
When to choose a carrier following criteria should be considered:
- High water solubility-to improve wet ability and enhance dissolution
- High glass transition point-improve stability
- Minimal water uptake
- Soluble in common solvent with drug-solvent evaporation
- Relatively low melting point-melting process
- Capable of forming a solid solution with the drug-similar solubility parameters
Comparative studies of different drugs:
Preliminary solubility studies of Sirolimus:
The solubility of Sirolimus in water at 25’c is 2.6 ug/ml. under the preence of mannitol, PEG 6000,Urea,PVP K-30,HPMC 5 pcs and beta- CD , the phase diagram of the sirolimus was taken out. The increasing of the correct amounts of dissolved drug was approximately linear for Mannitol, PEG -6000,Poloxamer-188 and beta-cyclodextrin.(Amidon and Crison,1995)
FTIR studies:
Solid dispersion of sirolimus and the pure drug of sirolimuswesr researched for FTIR spectral analysis. There was no any visible or non visible characteristic peaks for sirolimus in the solid dispersion.Hence it described that there is no interaction between the pure drug and the carrier used.
Dissolution testing:
The data from dissolution testing shows rapid dissolution of sirolimus from the solid dispersion and the solid complex when compared with the pure drug and the physical mixture. As the proportion of the poloxamer-188 and Mannitol carrier in the solid dispersion becomes higher the cumulative percentage of sirolimus. Solid dispersion prepared from solvent evaporation method with PEG -6000 in the ratio of 1:1 increased release with t50% of 60 minutes and achieved the highest amount of release of 99.45%
Enhancement of dissolution of Fenofibrate by solid dispersion technique:
Fenofibrate is a lipid lowering drug used in the treatment of hyperlipidemia, which is not soluble in water and lower absorption in gastric fluid. In order to increase the solubility and oral absorption of the drug in gastric fluid and to increase its dissolution rate solid dispersions and Lyophilization of dispersion is designed and evaluated. Solid dispersions of Fenofibrate were made using PEG 6000, Poloxamer 407 and a mixture of PEG 6000 and Poloxamer 407(1:1 mixture). The result of melt and solvent methods of making of solid dispersion on dissolution behavior was also invented.Dissolution studies showed a significant raise in dissolution of Fenofibrate when dispersed in PEG6000 and Poloxamer 407. Physical mixtures having PEG and Poloxamer 407 also indicated great dissolution of Fenofibrate as compared with that of pure drug, indicating the solubilizing effect of PEG6000 and Poloxamer 407.
Solid dispersions having Fenofibrate /Poloxamer 407, 1: 8, showed a 14-fold increase in dissolution after 60 min (D60) (Y.E and Wilken,1980) and another dispersion having Fenofibrate /PEG 6000, 1:10, showed an 8-folds. The dispersion having six parts of the PEG 6000: Poloxamer 407 mixture (PEG 4000/PEG 6000, 1:1 mixture) showed a 12-fold raise in D60 as compared with pure drug. When multi-carrier solid dispersion having six parts of mixture was prepared by the solvent method, the D60 value was about 2-fold that of the same dispersion made by the melt process. The dissolution of lyophilized solid dispersions further raised the dissolution of Fenofibratespecificly.(Sugimoto et al,1998)
Experimental methods:
Apparatus and chemicals:
Fenofibrate (99% purity) was obtained from Aurobindo pharmaceuticals, India. MCC, pregelatinized starch was obtained from Kemphasol, Bombay. PEG 6000 was purchased from Merck. Other excipients used were of analytical grade. (Hncock and Zografi,199
Preparation of solid dispersion of Fenofibrate:
Solvent Evaporation method using fluid-bed coating technique:
The Fenofibrate was dispersed in hydrophilic polymers like PEG 6000.Surfactants like PEG400 or Miglyol or Cremophore EL are needed to disperse Fenofibrate in polymer. Then this dispersion was dissolved with the help of IPA& DCM solvents. The solid solution of Fenofibrate polymer was coated onto inert bed of Sugar spheres or Lactose monohydrate using Fluid-bed coater granulater.Therefore solvent will be evaporated leaving a Fenofibrate-polymer coat surrounding the inert carrier. The dry mass will be achieved in granular state which was passed through 30 mesh (ACTM No. 30) and composition of solid dispersion.
Differential scanning calorimetry (DSC) analysis:
Thermal properties of Fenofibrate, polymer and solid dispersion were invented by using differential scanning calorimeter thermal analysis controller with an intracooler-2 cooling.
About 3 to 5 mg of product was taken in perforated aluminum sealed 50-μ l pans, and the heat runs for each sample was set from 40°C to 250°C at 10°C/min,(Munoz et al,1994) under an inert surroundings with the help of nitrogen. The apparatus was calibrated using pure metals like indium with known melting points and heat of fusion.
Dissolution studies:
Dissolution studies were taken in phosphate buffer (pH 7.2, 900 ml) at 37 ± 0.5 °C, using USP apparatus with a paddle rotating at 75 rpm.The samples equivalent to 120 mg fenofibrate, were concerned to dissolution. At fixed time difference samples (5 ml) were withdrawn and equal amount of fresh dissolution medium was added. Withdrawn samples were filtered through 0.45 μm membrane filter, and spectrophotometrically assayed for drug content at 287.4 nm wavelengths with the help of a UV-VIS spectrophotometer.
Stability studies:
Stability studies were taken out by keeping the samples in screw cap vials in stability chambers at 25 ± 2°C and 60 ± 5 % relative humidity (RH). Samples were taken regularly and researched for drug content and dissolution studies until 6 months. All the data are collected from the stability study were analyzed for significant differences by one-way Analysis.
Comparative studies of different drugs
Various Fenofibrate solid dispersions were made by using PEG-400,6000, MCC,pregelatinized starch in the mixture and individually, as taken by solvent evaporation technique to higher the solubility as well as dissolution of poorly aqueous soluble drug Fenofibrate. The DSC thermograms of pure fenofibrate and fenofibrate solid dispersion by using PEG-600.(McKenzie,1976)
Enhancement of dissolution of Furosemide by solid dispersion technique:
Materials and methods:
Materials:
The samples of FUR, PEG 6000, and PVP K30 (average molecular weights of 6000 and 50000, respectively ) were generous gifts from Maan Pharmaceuticals Ltd. (Mehsana, India) and were used without further purification . Directly compressible lactose, colloidal silicon dioxide, and magnesium stearate were produced from S.D Fine-Chem Ltd, Mumbai. All chemicals and solvents were of analytical reagent grade that had been used in the study. Throughout the work freshly distilled water was used. (Habib and Attia,1985)
Phase solubility study:
Phase-solubility research have been performed in line with the method noted through Higuchi and Connors (18). FUR, in amounts that exceeded its solubility, have been transferred to screw-capped vials made up of 30 mL aqueous PEG 6000 or maybe PVP K30 alternatives regarding diverse concentrations of mit (0, 1, 5, and 10%). The contents have been stirred while on an electromagnetic stirrer (Remi, India) at 25 °C and 37 °C for 72 h and 300 rpm.
This particular duration had been screened to be enough to reach equilibrium, after which no advancement in solubility was noticed. Immediately after hitting equilibrium, biological samples have been television through a 0. 22-µm membrane layer filtration system, very well diluted with 0. 1 And NaOH, and analyzed for drug content at the λmax regarding 274 nm (17) by using a spectrophotometer (Shimazdu-1601, UV–vis spectrophotometer, Shimadzu Corp, Kyoto, Japan). Most assays have been performed in triplicate.(Patel,2007)
Preparation of Solid Dispersion and Physical Mixture:
Solid Dispersions Served by Solvent Evaporation SDs involving of SDs within PEG 6000 as well as PVP K30 that contain distinct pounds rates (1: 1, 1: 5, 1: 10 in addition to denoted since SEPEG or SEPVP 1/1, 1/5, 1/10, respectively) were being made by the solvent technique (19) as follows.Toa answer involving FUR within ethanol (10 mg/25 mL), the right volume of PEG 6000 or PVP K30 seemed to be included. Solvent seemed to be evaporated under diminished pressure at 40 °C, and the caused deposit seemed to be dried out underneath vacuum cleaner intended for 3h, stored in a desiccator at least overnight, ground in a mortar, and passed through a 100 sieve.(Franco et al,2001)
Solid Dispersions Prepared by Melting of the Carrier:
Four SD preparations containing different weight ratios of FUR in PEG 6000 (1:1, 1:5, 1:10 and denoted as MEPEG 1/1, 1/5, 1/10, respectively) were prepared by the melting method (20). FUR was added to the melted PEG 6000 at 75 °C, and the resulting homogeneous preparation was rapidly cooled in a freezing mixture of ice and sodium chloride, and stored in a desiccator for 24 h. Subsequently, the dispersion was ground in a mortar and sieved through a 100 sieve.
Physical Mixtures
Physical mixtures (PMs) having the same weight ratios, as described in the previous two methods, were prepared by thoroughly mixing appropriate amounts of FUR and PEG 6000 or PVP K30 in a mortar until a homogeneous mixture was obtained. The resulting mixtures were sieved through a #120 sieve and denoted as PMPVP or PMPEG, respectively.
Characterization of Solid Dispersion:
Infrared (IR) Spectroscopic Analysis:
FTIR spectra of moisture-free powdered samples of FUR and its PMs and SDs with PEG 6000 and PVP K30 were obtained using a spectrophotometer (FTIR-8300, Shimadzu Co., Kyoto, Japan) by potassium bromide (KBr) pellet method. The scanning range was 750–4000 cm−1, and the resolution was 1 cm−1.(Van den Mooter,1998)
Powder X-ray Diffraction (PXRD) Analysis:
The physical state of FUR in the various preparations was evaluated by powder X-ray diffraction study. Powder X-ray diffraction patterns of all samples were determined using a Phillips PW 3710 scanner, IW 1830 generator with a CuK α anode at 40 kV and 30 mA, and a scan rate of 1° min−1 from 2θ range 1 to 40°.
Differential Scanning Calorimetry (DSC) Analysis:
DSC tests of most powdered biological samples were registered utilizing Shimadzu DSC-60 using TDA tendency series software program. All biological samples were weighed (8–10 mg) along with warmed up in a scanning rate associated with 10 °C/min under dry nitrogen flow (100 mL/min) between 50 and 300 °C. Aluminum pans along with lids were used for almost all biological samples. Pure water along with indium seeing that primary regular were used to calibrate the DSC temperature scale along with enthalpic reaction.
Wettability and Dissolution Studies:
A new wettability research was executed employing open up tubes comprising FUR and its PMs as well as SDs together with PEG 6000 as well as PVP K30; these were placed using lower capillary ends dropped into hued normal water (0. 01% eosin with water). The upward migration of the colored front was registered as a function of time (21). . Dissolution research involving FUR with powdered ingredients type and its PMs as well as SDs together with PEG 6000 as well as PVP K30 were executed to evaluate in vitro drug release profile.
Dissolution research were executed employing USP Apparatus 2 together with 500 mL dissolution medium (demineralized normal water comprising 0. 25% [w/v] involving salt lauryl sulfate [SLS]) at 37 ± 0. 5 °C (Teresa and Victoria,2002) as well as 50 rpm intended for 4h. Samples of pure FUR and PMs and SDs equivalent to 20 mg of the drug were added to the dissolution medium. At fixed time intervals, 5-mL aliquots were withdrawn, filtered through a 0.22-µm membrane filter, suitably diluted, and assayed for FUR content by measuring the absorbance at 274 nm using a spectropho- tometer. Equal volume of fresh medium prewarmed at the same temperature was replaced in the dissolution medium after each sampling to maintain constant volume throughout the test. Each test was performed in triplicate, and release curves were plotted using calculated mean values of cumulative drug release. Similarity factor (f2) and mean dissolution time (MDT) values were calculated to compare the extent of improvement in the dissolution rate of FUR from different samples.
Preliminary tests demonstrated that there was no change in the λmax of FUR due to the presence of PEG 6000 or PVP K30 dissolved in the dissolution medium.
Formulation Studies:
Formulation excipients were selected on the basis of preliminary tests, which demonstrated no interference of these excipients with the λmax of FUR. Tablets containing 20 mg of FUR were made by direct compression using different formulation excipients such as directly compressible lactose, colloidal silicon dioxide, and magnesium stearate. Tablets containing SDs equivalent to 20 mg FUR were made similarly. The blend was compressed on an eightstation single rotary machine (Cadmach, India) using round-shaped, flat punches to obtain tablets of 3–6 kg/cm2 hardness and 3.8–4.0 mm thickness. For the assay, three tablets were crushed, and a blend equivalent to 10 mg of FUR was weighed and dissolved in dissolution medium. The release profile of drug from tablets was studied in triplicate using the same dissolution media, conditions, and procedure as described for in vitro dissolution studies.
Comparative studies of different drugs
Phase-Solubility Study The solubility of FUR in water at 25 °C is 10 µg/mL; therefore, FUR can be considered to be a water-insoluble drug. The phase solubility curve of FUR in the presence of PEG and PVP at 25 and 37 °C . (For ease in discussion, hereafter, PEG 6000 and PVP K30 are abbreviated as PEG and PVP, respectively). From this curve, it can be seen that the apparent solubility of FUR increased with increasing temperature and carrier concentrations. At the highest polymer concentration (10% w/w), the solubility increased approximately 27-fold and 23-fold for PEG and PVP, respectively, at 37 °C. The same tendency was observed at 25 °C. An indication of the process of transfer of FUR from pure water to aqueous solution of PEG or PVP was obtained from the values of Gibbs free energy change (25). The Gibbs free energy of transfer (∆Gtr°) of FUR from pure water to aqueous solutions of SDs was calculated using the following equation:
Equation:
Where Sc/So is the ratio of molar solubility of FUR in aqueous solution of PEG or PVP to that of pure water. The enthalpy of transfer (∆Ht°) can be calculated from a modification of the van’t Hoff equation: ∆HRSS Tt coo =− () ( ) dln d1. The obtained values of ∆Gtr°, ∆Ht°, and apparent stability constants (Ka). The ∆Gtr° values show whether the reaction condition is favorable or unfavorable for drug solubilization in the aqueous carrier solution. Negative ∆Gtr° values indicate favorable conditions. ∆Gtr° and ∆Ht° values were all negative for both polymers at various concentrations, indicating the spontaneous nature of FUR solubilization, and decreased with an increase in PEG or PVP concentration, demonstrating that the reaction became more favorable as the concentration of PEG or PVP increased. These values also indicated that the extent of improvement in solubility was more with PEG as compared with PVP.
Characterization of SDs:
Fourier Transform Infrared (FTIR) Spectroscopic Analysis:
FTIR has been used to assess the interaction between carrier and guest molecules in the solid state. In the SD preparations, there is a peak band shift in the absorption spectrum of the guest. However, some of the changes are very subtle requiring careful interpretation of the spectrum.
The spectrum of pure FUR presented characteristic peaks at 3340 cm−1 (NH2 stretching vibration of Ar-NHCH2), 3260 cm−1 (stretching vibration of SO2NH2), 1665 cm−1 (bending vibration of amino group), 1560 cm−1 (asymmetric stretching vibration of the carboxyl group), and 1318 cm−1 (asymmetric stretching vibration of the sulfonyl group).
Important vibrations detected in the spectrum of PEG are the C–H stretching at 2890 cm−1 and the C–O (ether) stretching at 1125 cm−1. The spectrum of PVP showed important bands at 2925 cm−1 (C–H stretch) and 1652 cm−1 (C=O). A very broad band was also visible at 3300 cm−1, which was attributed to the presence of water confirming the broad endotherm detected in the DSC experiments.
The spectra of PMPEG 1/10 and PMPVP 1/10 can be simply regarded as the superposition of those of FUR and PEG or PVP. No difference was seen in the position of the absorption bands of FUR and PEG or PVP.
In the spectra of SEPEG 1/10, MEPEG 1/10, and SEPVP 1/10, the characteristic peaks of PEG or PVP were present at almost the same positions, whereas peaks due to FUR were absent indicating trapping of FUR inside the PEG or PVP matrix. Moreover, all the spectra showed no peaks other than those assigned to FUR, PEG, and PVP, which indicates the absence of any well-defined chemical interactions. Although hydrogen bonding between the hydrogen atom of the OH of the drug and oxygen atom in PEG or PVP could be expected, this was not demonstrated.
The presence of numerous distinct peaks in the PXRD spectrum indicate that FUR was present as a crystalline material with major characteristic diffraction peaks appearing at a diffraction angle of 2θ at 5.95, 11.98, 14.11, 18.05, 18.90, 20.36, 21.28, 22.82, 24.73, 27.48, and 29.17. PEG also exhibited a distinct pattern with diffraction peaks at 2θ at 15.00, 18.75, 23.15, 26.60, and 29.35, but the spectrum of PVP was characterized by the complete absence of any diffraction peak, which is characteristic of an amorphous compound.
The diffraction patterns of all the samples of SDs show peaks due to PEG or similar to PVP and an absence of major diffraction peaks corresponding to FUR, with most of the diffraction indicating FUR was present as amorphous material inside the PEG or PVP matrix. Moreover, no peaks other than those that could be assigned to pure FUR and PEG or PVP were detected in the SEPEG 1/10, MEPEG 1/10, and SEPVP 1/10, indicating no chemical interaction in the solid state between the two entities. In the case of physical mixing, diffractograms of PMPEG 1/10 showed more resemblance to PEG, whereas diffractograms of PMPVP 1/10 showed resemblance to FUR due to presence of free drug.
Differential Scanning Calorimetry (DSC) Studies:
DSC enables the quantitative detection of all processes in which energy is required or produced (i.e., endothermic or exothermic phase transformations). The thermal behavior of the prepared solid dispersions of FUR with PEG and PVP was studied by DSC. The FUR showed a melting peak at 225 °C with an enthalpy of fusion (∆H) of 302.22 mJ/g (26). The DSC scan of PVP showed a broad endotherm ranging from 80 to 120 °C due to the presence of residual moisture in PVP, whereas PEG showed a single sharp endotherm at 58 °C due to melting. DSC thermograms of PMPEG 1/10 and PMPVP 1/10 showed the melting peak of the drug at 225 °C, a sharp endothermic peak at 58 °C due to melting of PEG, and the broad endotherm due to the presence of water ranging from 90 to 110 °C in PVP. The DSC scans of SEPEG 1/10 and MEPEG 1/10 showed only one peak at 58 °C due to melting point of PEG, and the scan of SEPVP 1/10 showed one peak at 90–110 °C due to loss of water from PVP.
All samples of SDs showed complete absence of drug peak at 225 °C. This complete absence of the FUR peak indicates that FUR is amorphous or is in a solid solution inside the PEG and PVP matrix. This type of interaction was also observed in the FTIR and PXRD studies.
Wettability and Dissolution Studies:
The wettability regarding FUR has been appreciably increased simply by organizing it’s solid dispersions having PEG and PVP (Figure 5). The best progress regarding wettability in water has been witnessed having SEPEG 1/10 and SEPVP 1/10 (58. 7% and 49. 9%, respectively after 60 min). A large progress inside the wettability regarding FUR has been furthermore observed in PMPEG 1/10 and PMPVP 1/10 as compared having pure FUR (20%) after 60 min.
It really is typically accepted which dissolution advertising are not absolutely adviser regarding gastrointestinal (GI) disorders yet it is proposed in guidelines that a good method will employ a dissolution medium that is certainly physiologically purposeful or perhaps directly mimics in vivo disorders (27). It is often suggested which such as surface-active providers in dissolution advertising is significant with regard to inadequately soluble materials, because the lack of any area strain reducing realtor would lead to lesser wetting and in vitro dissolution rates which might be not necessarily adviser regarding in vivo conditions (27). It has been suggested that including surface-active agents in dissolution media is important for poorly soluble compounds, because the lack of a surface tension lowering agent would result in poorer wetting and in vitro dissolution rates that are not representative of in vivo rates (28). The FDA has permitted the use of surfactants in media for conducting dissolution studies of poorly soluble compounds (29).
Dissolution of pure FUR and all other prepared systems (SDs and PMs) were carried out in demineralized water containing 0.25% (w/v) SLS. DP30 min values (percent drug dissolved within 30 min), t50% (time to dissolve 50% drug), and mean dissolution time (MDT) values for different samples.In vitro dissolution profiles of pure FUR, its PM and SDs with PEG and PVP over a period of 4.
It is evident that the dissolution rate of pure FUR is very low (DP30 min 7.6%, t50% >> 4 h, and MDT of 58.3 min at 4 h). SDs of FUR with PEG and PVP significantly enhance the dissolution rate of FUR (80–95%, respectively) within 4 h as compared with PM as well as pure FUR. PMs with PEG and PVP also improved the dissolution rate of FUR. The highest improvement was obtained in SDs prepared with PEG by solvent evaporation techniques.
SEPEG 1/10 (97%) has a higher dissolution rate as compared with SEPVP 1/10 (88%) at the end of 4 hrs.
The obtained values of MDT for all samples are presented in Table 2. The MDT of pure FUR is very high (58.3 min). This value decreased to a greater extent after preparing its SDs and PM with PEG and PVP. SEPEG 1/10 showed the lowest MDT (20.2 min). MDT values of SDs prepared with PEG were lower than that with PVP. The same relationship was also observed with PM prepared with PEG and PVP also.
Comparisons between the release profiles of FUR from different samples were made by similarity factor f2. From this table, it is evident that the release profile of FUR from all the samples (i.e., SDs and PMs of PEG and PVP) and from pure FUR was dissimilar since f2 values for all these comparisons were less than 50. Release profiles of FUR from SEPEG and MEPEG at different concentrations were similar. Release of FUR from SDs with PEG and PVP were also significantly different from PMs with PEG and PVP at different concentration levels.
Formulation Studies:
The physical properties of all samples were studied to judge tabletting ability. In general, compressibility index values up to 15% and an angle of repose between 25 and 30 results in good to excellent flow properties (30). Percentage compressibility and the angle of repose of samples are. These values indicate good compressibility and flow properties, making these samples suitable for tableting.
Release profiles of FUR from conventional tablets containing FUR (without PEG or PVP) and tablets containing SDs and PMs of FUR with PEG or PVP Are shown.Release of FUR from tablets containing SDs with PVP or PEG was faster and greater as compared with conventional tablets containing FUR. This confirmed the advantage of improved aqueous solubility of FUR in its SD form, which can be formulated as tablets with better dissolution characteristics.
DP30min, t50%, and MDT values for release of FUR from tablets prepared using different samples. DP30min values were higher for tablets prepared using SDs and PMs as compared with those of conventional tablets containing only FUR (2.9), whereas t50% and MDT values of FUR from tablets containing SDs and PMs were significantly lower than those of conventional tablets containing only FUR and no PEG or PVP (76.0 min and >4 h, respectively).
DISCUSSION
After studying about the various method of solid dispersion, we can say that there are some methods which are followed by individual drugs. In recent days these three methods are widely used in these following drugs.
- Physical mixing method
- Solid evaporation method
- Melting method
The drugs that follow these three methods are-
- .Fenofibrate
- .Furosemide
- .Sirolimus
- .Lurasidone HCL
- .Terbinafine hydrochloride
- .Indomethacin
- .Simvastatin
- .Meloxicam
- .Gliclazide
- .Lornoxicam
- .Diacerein
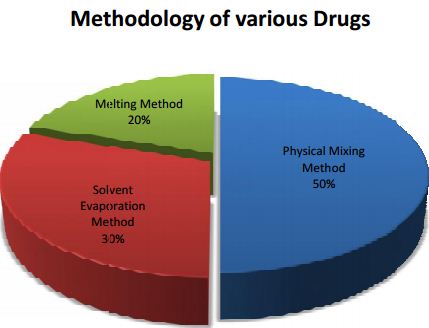
From this Pi chart we can say that, 50% of the drugs are followed by physical mixing method 3Lurasidone HCl, Terbinafine, Indomethacin, Simvastatin, Meloxicam, Gliclazide, Diacerein are dispersed by using physical mixing method.
In this kneading method, transporter is permeated with water and transformed to paste. Drug is then added and kneaded for particular time. The kneaded amalgamation is then dried and passed through sieve if obligatory.0% by solvent evaporation method and remaining 20% by the melting method.
Conclusion:
Solid dispersion has great possible both for increasing the bioavailability of drug and mounting controlled release preparations. in regard to manufacturing considerations the problem of total solvent removal in dispersions prepared by solvent evaporation way need to be addressed (Singh et al,1966). With particle-coating utensils new commercially available, this course has a promising future. The problem of volatility of the supersaturated state upon dissolution, which results in a stable form, has been dealt with by addition of a retarding agent. Methylcellulose used as a retarding agent in dispersions of indomethacin and flufenamic acid in PVP (SjokvistE,1992). Proscribed release formulations of acetaminophen, aminopyrine, chlorpheniramine maleate and salicylic acid that use eudragit RS as water insoluble carrier organized by solvent method, have been reported. Valuable preliminary studies of the use of solid dispersions to provide sustained release or controlled release of drugs have been reported.(Slade L,1991)
Solid dispersion can boost dissolution rate of drugs with poor water solubility of these systems requirements consideration. Physical and chemical stability of both the drug and the transporterin a solid dispersion are major developmental issues,as exemplified by the recent withdrawal of ritonavir capsules from the market, So future research desires to be directed to address diverse stability issues. solid dispersions can progress their stability and recital by increasing drug-polymer solubility, amorphous fraction, particle wettability and particle porosity. Moreover, new, optimized manufacturing techniques that are easily scalable are also coming out of academic and industrial research. Further studies on scale up and validation of the process will essential.