Sickle cell disease (Symptoms, Diagnosis, and Treatment)
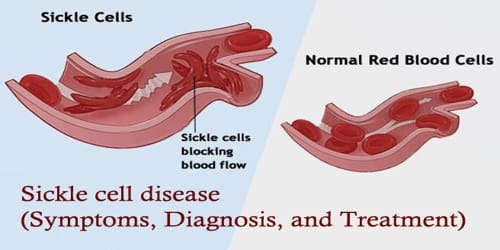
Definition: Sickle cell disease (SCD) or Sickle cell is a genetic disease of the red blood cells (RBCs). Normally, RBCs are shaped like a disc, which gives them the flexibility to travel through even the smallest blood vessels. This leads to a rigid, sickle-like shape under certain circumstances. Problems in sickle cell disease typically begin around 5 to 6 months of age. People with this disorder have atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape.
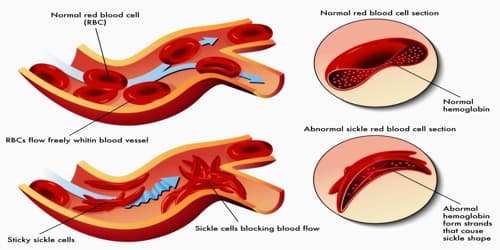
Sickle cell disease occurs when a person inherits two abnormal copies of the hemoglobin gene, one from each parent. This gene occurs in chromosome 11. Several subtypes exist, depending on the exact mutation in each hemoglobin gene.
The condition was first described in the medical literature by the American physician James B. Herrick in 1910. In 1949, the genetic transmission was determined by E. A. Beet and J. V. Neel. In 1954, the protective effect against malaria of sickle cell trait was described.
Other names of this condition:
- HbS disease
- Hemoglobin S Disease
- SCD
- Sickle cell disorders
- Sickling disorder due to hemoglobin S
The signs and symptoms of sickle cell disease are caused by the sickling of red blood cells. When red blood cells sickle, they break down prematurely, which can lead to anemia. Anemia can cause shortness of breath, fatigue, and delayed growth and development in children. The rapid breakdown of red blood cells may also cause yellowing of the eyes and skin, which are signs of jaundice. Painful episodes can occur when sickled red blood cells, which are stiff and inflexible, get stuck in small blood vessels.
Diagnosis is by a blood test, and some countries test all babies at birth for the disease. Diagnosis is also possible during pregnancy.
There’s no cure for most people with sickle cell anemia. But treatments can relieve pain and help prevent problems associated with the disease.
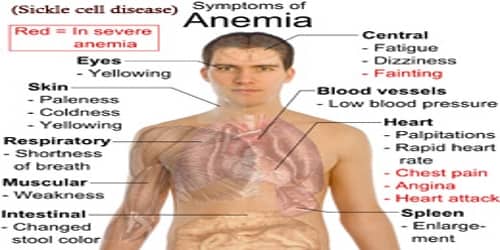
Signs and Symptoms of Sickle cell disease (SCD): Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person. Sickle cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.
While there are multiple types of SCD, they all have similar symptoms, which vary in severity. These include:
- excessive fatigue or irritability, from anemia
- fussiness, in babies
- bedwetting, from associated kidney problems
- jaundice, which is yellowing of the eyes and skin
- swelling and pain in hands and feet
- frequent infections
- pain in the chest, back, arms, or legs
For a baby to be born with sickle cell anemia, both parents must carry a sickle cell gene.
Diagnosis and Treatment of Sickle cell disease (SCD): A blood test can check for hemoglobin S the defective form of hemoglobin that underlies sickle cell anemia. This blood test is part of routine newborn screening done at the hospital. But older children and adults can be tested, too.
Several blood tests can be used to look for SCD:
- Blood counts can reveal an abnormal Hb level in the range of 6 to 8 grams per deciliter.
- Blood films may show RBCs that appear as irregularly contracted cells.
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle hemoglobin can also be demonstrated with the “sickle solubility test”. A mixture of hemoglobin S (Hb S) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.
Hb electrophoresis is always needed to confirm the diagnosis of sickle cell disease. It measures the different types of hemoglobin in the blood.
Sickle cell disease can be diagnosed in an unborn baby by sampling some of the fluid surrounding the baby in the mother’s womb (amniotic fluid) to look for the sickle cell gene.
Treatment involves a number of measures. L-glutamine use was supported by the FDA starting at the age of 5 as it decreases complications. Babies and children age 2 and younger with sickle cell anemia should make frequent visits to a doctor. Children older than 2 and adults with sickle cell anemia should see a doctor at least once a year.
Pain medication is used to relieve the pain during a sickle crisis. People may need over-the-counter drugs or strong prescription pain medication like morphine. (Droxia, Hydrea) helps to increase production of fetal hemoglobin. It may reduce the number of blood transfusions. Immunizations can help prevent infections. Patients tend to have lower immunity.
Treatments might include medications to reduce pain and prevent complications, and blood transfusions, as well as a bone marrow transplant.
Childhood vaccinations are important for preventing disease in all children. They’re even more important for children with sickle cell anemia because their infections can be severe. Vaccinations, such as the pneumococcal vaccine and the annual flu shot, are also important for adults with sickle cell anemia.
There are things people can do at home to help their sickle cell symptoms:
- Use heating pads for pain relief.
- Take folic acid supplements, as recommended by their doctor.
- Eat an adequate amount of fruits, vegetables, and whole-wheat grains. Doing so can help their body make more RBCs.
- Drink more water to reduce the chances of sickle cell crises.
- Exercise regularly and reduce stress to reduce crises, too.
Contact the doctor immediately if they think they have any type of infection. Early treatment of an infection may prevent a full-blown crisis.
Information Source: