Introduction:
For many years, pressurized metered dose inhalers (pMDIs) have been the leading application form of inhalation therapy. With their favorable features, such as easy handling, high reliability and accurate metering performance, pMDIs currently account for more than 90% of all inhaled asthma therapy. However, traditional pMDIs also have a number of important drawbacks; for example, they require difficult hand–lung coordination by the patient and use environmentally damaging CFC propellants. Although newer pMDIs and formulations, which go some way to addressing these problems, are gradually becoming available, this type of drug delivery is intrinsically inefficient and, in normal circumstances, only 10–15% of the dose reaches the lung. Moreover, formulating drugs for use in pMDIs is difficult, especially with the newer propellants. By contrast, nebulizers are very efficient at creating mists of extremely fine droplets with good pulmonary deposition. However, it can take several minutes for a patient to inhale a dose from a nebulizer and these devices have traditionally been used primarily in institutional environments because of their considerable size and operational complexity. Dry powder inhalers (DPIs) represent a significant advance in pulmonary delivery technology. For example, they are breath-actuated and so coordination problems such as synchronizing dose discharge with inhalation are overcome. DPIs are also potentially suitable for delivering a wider range of drugs than pMDIs, including biopharmaceutical systemic therapies such as peptides and proteins. Indeed, DPIs can deliver a range of doses from less than 10 mg to more than 20 mg via one short inhalation. There is a wide range of different types of DPI device already on the market or in development. The older devices (such as RotahalerÔ) discharge powder from a hard gelatin capsule inserted by the patient, but these are now being replaced by either multi-unit dose devices with blisters (such as DiskusÔ and SpirosÔ) or reservoir-type systems (such as TurbuhalerÔ and ClickhalerÔ).
Pharmaceutical inhalation aerosols offer a distinct advantage of rapid delivery of drugs to the site of action as exemplified traditionally by the administration of bronchodilators for asthma treatment. More recently, driven by the pharmaceutical industry to explore the potential of the lungs for systemic treatment of diseases, pulmonary drug delivery by inhalation aerosols has been undergoing rapid development [1]. Significant research and development efforts have been put into dry powder aerosols, which require no propellant, have superior chemical stability compared with solution, and are easy of use (hence good patient compliance). Successful development of a dry powder aerosol delivery system requires careful considerations of: (i) production of the drug powder; (ii) formulation of the powder; and (iii) aerosol generation and delivery by the inhaler device, since all of these factors will affect deposition of an aerosol in the lungs. Recent research and development addressing the above issues will be discussed in this review. It is hoped that this article will not only provide the latest information on dry powder aerosols to the colloid and surface scientists who are working in the medical and drug delivery fields, but also help to stimulate the interests of those who may consider moving into this research area.
Dry powder aerosol drug delivery system includes powder production, formulation, dispersion, and delivery of the powder aerosol to the lung. Conventional method of production of fine particles suitable for aerosol delivery involves crystallization and micronisation by milling. This method has many deficiencies including uncontrolled particle size distribution and amorphous materials. As a result, development of alternative methods such as supercritical fluid technology and high gravity controlled precipitation becomes important. Over the last decade performance of powder formulations has been improved significantly through the use of engineered drug particles and excipient systems which are: (i) of low aerodynamic diameters (using porous particles with low density) and/or (ii) less cohesive and adhesive (by reducing surface energy, using particles with corrugated surfaces, hydrophobic additives and fine carrier particles). Analytical techniques such as the atomic force microscopy (AFM) and inverse gas chromatography (IGC) have been used to probe particle forces and surface energy affecting powder dispersion. Relative humidity (RH) is critical to the performance of dry powder inhalers (DPI) via capillary force and electrostatic interaction. Electrostatic charge of different particle size fractions of an aerosol can now be measured using a modified electrical low-pressure impact or (ELPI). Compared with powders, much less work has been done on the inhaler devices at the fundamental level. Most recently, computational fluid dynamics has been applied to understand how the inhaler design (such as mouthpiece, grid structure, air inlet) affects powder dispersion. Surface and colloid science can be applied to each of these areas to benefit the research and development of dry powder aerosol drug delivery.
Characteristics of dry powder for pulmonary drag delivery:
A more recent approach to improve the aerosol performance is through the use of engineered particles that are: (i) of low aerodynamic diameters and/or (ii) less cohesive and adhesive , [1, 2, and 10]: Aerodynamic diameter (Da) is a key parameter determining whether an aerosol particle is inhalable. For a spherical particle, Da is simply equal to the physical diameter (D) times the square root of the particle density (ρ). Hence, at a given density, small particles have a lower aerodynamic diameter than the large particles. For example, three different sizes (3, 5 and 7 _m) small particles of mannitol were prepared by spray drying. Small size particles were obtained by using a lower mannitol concentration and/or a higher atomizing pressure. When the powders were dispersed by a commercial inhaler device (Dinkihaler®), a large amount of fine particles less than 5 _m in the aerosol cloud was achievable [11]. Similar results were reported for another anti-asthmatic drug, disodium cromoglycate [12]. Since porous particles contain a high void space, the particle density is low, leading to a low Da (based on Da =Dρ1/2). This approach has been successfully adopted by both Nektar Therapeutics and Alkermes Inc. in their dry powder formulation to enhance the aerosol performance. Porous particles like those shown in Fig. 2c have been prepared by spray drying of emulsions containing a highly volatile solvent or a blowing agent with a high vapour pressure. The pores were created as the solvent was rapidly evaporated from within the particle. However, the structural stability of these porous particles is unknown. Non-spherical particles such as elongated or needle-like objects have interesting aerodynamic properties. Da of elongated particles is controlled mainly by the short axis

rather than the long axis. It was found that Da increases with the aspect ratio (i.e. ratio of the long axis to the short axis) of an elongated particle. When the aspect ratio is greater than 15 or 20, Da will be about two to three times the short axis (physical diameter). Any further increase in the aspect ratio will only increase the Da very little. In other words, Da can be made small by having very long particles as long as the short axis remains small (e.g. ≤1_m). Elongated particles of anti-asthmatic drugs such as cromoglycic acid and nedocromil were prepared by controlled crystallization (via anti solvent precipitation or cooling from a saturated aqueous solution) followed by spray drying to obtain the powder [13, 14]. Solid particles with corrugated (rough) surface have also been successfully used to improve the aerosol performance. Bovine serum albumin was used as a model protein drug and particles with different degree of surface corrugation were prepared by spray drying. Increased corrugation was obtained using a low protein concentration (<50 mg/ml) along with a low atomization rate of <600 L/h [15, 16]. These particles are likely to result in larger inter-particulate separation and less contact area, both of which will cause reduced Vander Waals force of attraction. The degree of roughness was quantified by surface fractal dimension [17, 18], and only a small degree of surface roughness is required to increase the aerosol performance of the powder [16]. Powder cohesion and dispersion will depend on the surface energy, which has been determined for a number of pharmaceutical powders using inverse gas chromatography (IGC). Correlation between the surface energy and the powder dispersion has been obtained for certain compounds such as albuterol sulfate, salmeterol xinafoate, and mannitol [19, 20]. Further correlations between a surface energy parameter and fine particle fraction were reported on albuterol and ipratropium using sugar carrier systems [21]. However, a correlation could not be found for disodium cromoglycate powders containing amino acids as an additive, indicating that other physico-chemical parameters may also be involved [22]. Since IGC is the major technique used in these studies for probing surface energy, other surface analytical methods such as contact angle measurements or organic vapour uptake could be undertaken to provide additional information on the surface energy. Moisture is well known to affect powder cohesion (and dispersion as aerosol) through capillary force at high relative humidity (RH) while low RH favors electrostatic interaction. Fundamental study of the effects of RH on adhesion due to capillary force has been undertaken by atomic force microscopy [23], although translating the single particle results of AFM to the bulk powder situation involving multiple particle interactions in the dry powder inhalers (DPI) is not straight-forward.
Dry powder Design and computational simulation for pulmonary drug delivery:
The performance of a powder aerosol delivery system depends not only on the powder formulation but also the inhaler device [1]. However, much less work has been done at a fundamental level on inhalers compared with powders. All of the marketed inhalers are ‘passive’ devices, relying on the inspiratory air-flow of the patient to disperse the powder formulation into individual particles or smaller agglomerates for inhalation. However, concepts on how the powder interacts with the device during dispersion are generally vague (e.g. turbulence is thought to be the principal force for deaggregation but other factors are also involved) [33].
Air turbulence and mechanical impaction (particle–particle, particle–device) are generally accepted as mechanisms controlling powder dispersion in the device (Fig. 3), but details of their relative importance are much less clear [34]. In addition, a large number of these devices use a capsule to store and dispense the drug formulation, but a fundamental understanding of the role of capsule and the influence of air-flow is lacking. In a recent text Finlay discussed some of the important aspects for turbulent deaggregation of agglomerates in dry powder inhalers [35]. It was pointed out that the turbulence scales need to be considered and the integral scale strain rate (which is a measure of the velocity gradient across the integral scale eddies and the most energetic occurring in a turbulent flow) is a more appropriate parameter to study agglomerate break-up. By applying computational fluid dynamics we have begun to understand more about air-flow and deagglomeration in inhaler devices. It was demonstrated that small variations in the device design can produce significant performance variations in a dry powder inhaler [36]. Computational results showed that compared with the Rotahaler®, the Aeroliser® generates a stronger flow field with significantly greater (approximately three times) turbulence (local eddies) levels and local eddies as well as a greater number of particle–device impactions for break-up of agglomerates. A grid is present in both inhalers. The grid in the Rotahaler® acts to generate turbulence and to slow down the flow exiting the device whereas the one in the Aeroliser® also acts to straighten the flow out of the mouthpiece. Furthermore, a more open grid has lower eddies than the full (original) grid at a plane 2mm upstream from the grid. The effects of mouthpiece length, inlet air dimension, presence of a capsule and its size, and air-flow rate have also been studied for the Aeroliser® [37, 38]. While CFD is able to track single particle motion in the device and to provide the turbulence level as well as impaction information, exactly how agglomerate breaks up during impaction and interaction with the air turbulence would require additional modelling techniques such as the discrete element method (DEM). DEM has been employed in other powder areas [39–41] and would enhance our understanding of powder packing and agglomerate breakage in the DPI systems.
Development of dry powder for enhancing pulmonary delivery:
Dry powder inhalers:
The aerosolization or inhalation of medicaments by humans has been used since late the 1950s and since 1956, the pressurized metered dose inhaler (pMDI) become the most commonly used device to deliver inhaled asthma drugs (Freedman, 1956); however, with the advancement of science and technology, pulmonary delivery of drugs has become the route of choice after the introduction of the DPI in 1967 (Altounyan, 1967; Bell et al., 1971). Inhalation therapy, or pulmonary drug delivery, via pMDIs, DPIs or nebulisers, is the preferredmethod of treating patients with asthma (Kirk, 1986; Byron and Patton, 1994; Sheth, 2002). The clinical features of nebulisers, pMDIs and DPIs have recently been compared and pulmonary drug delivery is increasingly becoming a target for systemic drug delivery as a result of its inherent convenience, ability to administer drugs with poor oral availability, and the large surface area of lungs and long residence times associated with peripheral lung deposition (Byron and Patton, 1994; Patton, 1996; Cochraneet al., 2000; Groneberg et al., 2003). DPIs represent the most rapidly expanding field in pulmonary drug delivery in recent years, largely as a result of the perceived limitations in pMDIs and nebulisers (Hickey et al., 1994). Unlike pMDIs, DPIs avoid problems inherent in the use of propellant gases and the need for coordination of inhalation and actuation (Hickey et al., 1994). DPIs are also very portable, patient friendly, easy to use and do not require spacers (Geller, 2005). DPIs are subject to strict pharmaceutical and manufacturing standards by regulatory bodies, the most challenging of which is the demonstration of device reliability in terms of delivered dose uniformity.
Development of an ideal DPI:
Dry powders were made with DPPC, albumin and a sugar (lactose or trehalose) or a The inhalation device is important in achieving adequate delivery of inhaled drug to lungs. The device should be easy to use, inexpensive and portable. The device must provide an environment where the drug can maintain its physicochemical stability and produce reproducible drug dosing. The device should be designed to deliver high fine particle fraction (FPF) of drugs from the formulations (Srichana et al., 1998). However, devices with higher resistance need a higher inspiratory force by the patients to achieve the desired air flow. This could be difficult for patients with severe asthma and for children and infants. Therefore, a balance between these two factors is necessary to achieve the desired therapeutic effect from DPI formulations. For an ideal DPI a number of characteristics are important for device reliability, clinical efficacy and patient acceptance. These include:
• A device which is simple to use, convenient to carry, contains multiple doses, protects the drug
from moisture and has a indicator (audiovisual) of doses remaining (Ashurst et al., 2000; Sheth,
2002; Helen, 2001; Newman, 2004; Hickey and Crowder, 2007);
• Dose delivery which is accurate and uniform over awide range of inspiratoryflowrates (Ashurst
et al., 2000; Sheth, 2002; Newman, 2004; Chrystyn, 2006);
• Consistent dose delivery throughout the life of the inhaler and consistency of dose when
compared to other similar inhalers (Ashurst et al., 2000; Newman and Busse, 2002);
• Optimal particle size of drug for deep lung delivery (Clark, 1995);
• Suitability for a wide range of drugs and doses (Newman, 2004);
• Minimum adhesion between drug formulation and devices (Byron, 2004);
• Product stability in the device(Clark, 1995; Ashurst et al., 2000; Byron, 2004; Newman, 2004);
• Cost-effectiveness (Clark, 1995; Bisgaard, 1996); and
• Feedback mechanism to inform the patient of dose administration (Ashurst et al., 2000;
Newman, 2004).
No DPIs achieve all of these ideal characteristics; however, considerable research is being conducted to improve their performance characteristics where necessary. Some of these ideal characteristics are more important than others and will require different levels of improvement and/or innovation. Furthermore, others are influenced by the need for patient education in the proper use and storage of their DPI.
Considerations in the development of DPIs:
For DPIs and other inhalers the dose received by the patient is dependent on four interrelated factors (Atkins, 2005; Hess, 2005; Chan, 2006):
1. the properties of the drug formulation, particularly powder flow, particle size and drug–carrier interaction;
2. the performance of the inhaler device, including aerosol generation and delivery;
3. correct inhalation technique for deposition in the lungs; and
4.the inspiratory flow rate.
The optimisation of the drug formulation is often dependent upon the type of device used and as such they are often formulated together (Newman and Busse, 2002; Atkins, 2005). Therefore, the inhaler–drug combination is generally considered a unique medication whose in vitro performance and in vivo efficacy must be demonstrated (de Boer et al., 1996; Borgstrom et al., 2005; Sato et al., 2005; Rosenstock et al., 2007). For example, lung deposition of budesonide (1000_g) delivered via a Turbuhaler® was 2.2-fold higher than that of fluticasone propionate (1000_g) via Diskus®. The systemic bioavailability of budesonide via Turbuhaler® was 3-fold higher than fluticasone via Diskus®; however, similar plasma cortisol suppressionwas observed in both cases (Thorson et al., 2001). In another study, inhalation of fluticasone propionate (250_g) delivered via Diskus® inhaler and budesonide (600_g) delivered via Turbuhaler® in patients with asthma has been conducted and fluticasone propionate produced similar effect compared to that of budesonide (Backman et al., 2001). Higher lung deposition (31.0%) of budesonide (800_g) via a Turbuhaler® was also observed while compared with that of fluticasone (750_g) deposition (8%) via a Diskus® inhaler (Agertoft and Pederson, 2003). In vitro and in vivo performances of a new device Swinghaler®, a multi-dose inhaler device has been evaluated for procaterol and budesonide, and Swinghaler® showed an equivalent plasma concentration of budesonide as that using the Turbuhaler® (Sato et al., 2005). Using a new device both in vitro and in vivo delivery of a tailor made placebo powder and insulin, was carried out and more than 50% lung deposition of powders was observed (Rosenstock et al., 2007). There aremany factors that affect the quality of device, formulation, and drug delivery pattern from different devices. Therefore, well-defined in vitro and in vivo studies may help select best inhalers to achieve maximum therapeutic benefits. Clinical effectiveness of a DPI is also influenced by drug factors such as potency, pharmacokinetics, safety and efficacy, patient factors (such as disease severity and age), inhalation technique and compliance (Kelly, 2002). Similar levels of clinical effectiveness of different DPIs, particularly for asthma, have been demonstrated through randomised controlled trials. However, this is sometimes questioned in real-life where patient factors are more variable (Thomas and Williams, 2005). Despite this, patient factors can be harmonised to a certain extent, particularly factors such as inhalation technique and compliance. Evidence has shown that apatient’s pattern of inhalation can be changed with education so as to improve the performance of the DPI (Kelly, 2002; Smith and Parry-Billings, 2003; Hess, 2005).
Current dry powder inhalers on the market:
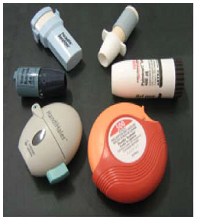
The inhaler device is very important in successful development of DPI products. Presently, over 20 DPI devices are available on the market and more than 25 are in development, but no device meets all of the requirements of an ideal DPI device mentioned in Section 2.1. A list of current DPI devices with delivery mechanism has been presented in Table 1. Photographs of some currently available devices are presented in Fig. 1. There is a wide range of DPI devices, single or multiple dose devices, breath activated and power driven, are available in the market (Table 1); however, the development of novel devices with new designs continues because the design of device affects DPI performance (Coates et al., 2004). Currently, based on the design, DPI devices may be classified into three broad categories, i.e., the first generation DPIs, the second generation DPIs and the third generation DPIs. The first generation DPIs were breath activated single unit dose (capsule), i.e., the Spinhaler® and Rotahaler® and the drug delivery issues were related to particle size and deagglomeration of drug–carrier agglomerates or drug–carrier mixtures delivered by patient’s inspiratory flow. The second generation of DPIs use better technology, i.e., multi-dose DPIs (they measure the dose from a powder reservoir) or multi-unit dose (they disperse individual doses which are premetered into blisters, disks, dimples, tubes and strip by the manufacturers) and multi-unit dose devices are likely to ensure the reproducibility of the formulation compared to that of multi-dose reservoir. All DPIs devices have some essential components incorporated with the device such as drug holder, the air inlet, the deagglomeration

The design of DPIs is developed in such away that the device should induce sufficient turbulence and particle–particle collisions to detached drug particles from the carrier surface (interactive mixtures) or deagglomerates particles from large agglomerates of drugs only. The majority of DPI devices are primed by pressing (Rotahaler®), sliding (Spinhaler®), rotating (Twisthaler®) or piercing (Handihaler®) to prepare the dose for fluidization with tangential flow of air during patient inspiration. The fluidised powder is then passed through a screen (incorporated within the device), which deagglomerates particles for deep lung delivery. Nevertheless, lung deposition from these inhalers varies from 12 to 40% (Steckel and Muller, 1997; Dunbar, 2002; Hickey, 2004; DiNunzio et al., 2007). The third generation DPIs, also known as active devices, which employ compressed gas or motor driven impellers or use electronic vibration (Crowder et al., 2001; Young et al., 2004; Brown et al., 2004) to disperse drug from the formulation. These devices are more sophisticated but user-friendly. Due to the presence of an energy source, active devices enable respiratory force independent dosing precision and reproducible aerosol production. The very first approved active device (Exubera®, Pfizer) with compressed air to aerosolise drug formulation for DPI insulin delivery was until recently available on market. This DPI with insulin was anticipated to be cost effective compared to that of insulin injection. However, this large and clumsy device has failed to achieve recognition of physicians and patients. While passive inhalation is commonly used in DPIs designed for topical respiratory drug delivery, active dispersion mechanisms (i.e., where the device inputs the energy) are considered desirable for drugs intended for systemic action which have to penetrate more deeply into the lungs (Schultz et al., 1992; Hil, 1994). The efficiency of breath actuated DPI devices depends on the patient’s inspiratory force, whereas, the powder dispersion from active DPIs is limited to the physical or electrical mechanism (vibration, compressed air, impact force and impellers available in the device (Crowder et al., 2001; Young et al., 2004; Brown et al., 2004); however, active DPIs are useful for aged people.
There are design differences in these devices including the presence of grids, baffles, constrictions, diameter and length of inhalation channel, positioning of mouthpiece, and orientation of inclination of device (Timsina et al., 1994). As mentioned before, single- or multi-unit dose devices have individual pre-metered doses sealed in the device, whereas in reservoir devices the patient dispenses the dose at each use. Single-dose and multi-unit dose inhalers are more effective than multi-dose reservoir devices as they ensure dose consistency and avoid the effects of moisture in the powder reservoir (Steckel and Muller, 1997). Dosing uniformity and storage stability is difficult when powders are delivered from a bulk powder. To ensure effective drug delivery into the lower airway of lungs the inspiratory flow rate must be sufficient to produce adequate turbulent air flow in any devices so that adequate aerosol cloud of the aerosolised fine particles. Therefore, a balance between the design of an inhaler device, drug formulation, and the inspiratory flow rate of patient is required (Steckel and Muller, 1997; Srichana et al., 1998). According to Ashurst et al. (2000), in order for new DPI designs to establish their own place in the market they should show advantages over existing devices. It is generally very difficult to compare devices because they often deliver different medications. Numerous research articles and review papers have been published demonstrating designs and performances of various devices and the readers are referred to those papers for further details (Newman, 2004; Chrystyn, 2006; Dalby et al., 2007). A large number of new devices with various designs and various types of drug delivery mechanisms have been developed; however, none of them showed superficial efficiency in delivering drugs into the deep lungs. Market is rapidly expanding and a large number of novel devices are in development with limited published data and some of them will be approved in the near future (Table 2).
Limitations in the reliability of existing DPIs:
The respiratory pattern of patients during aerosol intake may influence the deposition of inhaled particles, because the mean flow rates of particles in each region of the airways is governed by the breathing volume and frequency of breathing (Byron, 1986; Gonda, 1990; Martonen and Katz, 1993). The main limitation with existing DPIs is that delivery of the drug is often dependent upon inspiratory flowrates for effective delivery of the drug powder (Ganderton and Kasem, 1992; de Boer et al., 1996; Hickey and Concessio, 1997) and deagglomeration of drug particles (Lucas et al., 1998; Zeng et al., 1998; Louey and Stewart, 2002; Islam et al., 2004; Adi et al., 2006). For example, some DPIs require inspiratory flow of ≥30 L/min to effectively deagglomerate the powder (de Boer et al., 1996). However as discussed above, the breath actuation characteristic of DPIs is also thought to be one of their main strengths and drug delivery can also be influenced by the rate of increase in inspiratory air flow (Newman and Busse, 2002). The majority of passive DPIs are less efficient at lower flowrates (<30 L/min) so that optimal lung deposition will only occur if the patient is able to achieve a sufficiently rapid and deep inhalation (Sheth, 2002). Low-resistance passive DPIs are generally less dependent on flow rate than high-resistance devices. Inspiratory flow rate was found to play the most important role in determining the dispersion of salbutamol sulphate aerosolized from a Rotahaler® (Steckel and Muller, 1997; Srichana et al., 1998; Zeng et al., 2000). A flow rate of 60 L/min has been reported to be advantageous for effective delivery of drugs from Turbuhaler® (de Boer et al., 1996) and patients can achieve sufficient inspiratory effort to deagglomerate and aerosolise the dose (Li and Edwards, 1997). Increased inspiratory flow rate may help increase the deposition of particles in the upper airways. Slow inhalation rate increases the number of particles to reach in the peripheral region of the respiratory tract by impaction. A slow inhalation rate (25 L/min) with breath holding showed maximal deposition of terbutaline sulphate compared to the faster rate (80 L/min) of inhalation (Newman et al., 1981). However, some DPIs such as the Clickhaler® (Nantel et al., 1999) and the Easyhaler® (Palander et al., 2000; Tarsin et al., 2004) have showed uniform delivery of doses independent of flow rate compared to that of Turbuhaler® (Newhouse et al., 1999). Conversely, the active devices are designed to deliver drugs independent of the patient’s inspiration but powder dispersion is dependent on the physical or electrical mechanisms. Despite these well-recognised limitations, the importance of device reliability and drug deposition depends somewhat on the drug’s physicochemical properties and the clinical indication. There is some debate about the importance of better drug deposition in patients with asthma but little about the need for improved deposition deep into the lung for treatment of systemic disease. Despite this, device performance should be assessed over a range of flow rates to account for all possible patients and clinical circumstances.
Discussions and conclusion:
Despite appropriate standards of device reliability being a requirement for licensing and marketing of DPIs, there remain areas for improvement. Innovation and improvements in device reliability may not be mutually exclusive and neither is more important than the other. Until now, the cost of producing an inhaler economically, and with the necessary performance, has not been possible. In addition, there are many potential novel applications for DPIs for which device innovation will be necessary. The application of active DPI devices (the use of electrically driven dispersion) provides an opportunity to enhance the efficiency for the aged patients. According to the food and drug administration (FDA) it is recommended to add a necessary part like an integral dose counter as an active part of DPI device (CRER, 1998). With the advancing technology, the future DPI devices may add other features like dose reminder, audiovisual signals of dose delivery, measurement of flow rates during inhalation (Hickey and Crowder, 2007). Along with the device design, there is a great concern about the interaction between formulation and device that has to be accounted during designing a new device. The effect of DPI formulation (type of lactose and physicochemical properties of drug), capsule material and inhalers on the charge and polarity of DPI aerosols have been demonstrated and the type of the charge acquired by the particle was dependent on the type of inhaler, carrier particle size and capsule shell used for a formulation (Telko and Hickey, 2005). Therefore, these factors should be considered for an ideal inhaler which is more reliable, efficient, user friendly and cost effective. Most of the manufacturers and researchers are looking for novel efficient devices because in 2007, more than 20 new patent applications were filed for new designs of inhalers or parts of inhalers. Various studies have been conducted to compare the performance of DPIs; however, very limited number revealed insight into themechanism of drug dispersion fromthe devices. Drug delivery mechanism is important to rational design of efficient inhalerwith improved performance. With the changing technology, device development has progressed tremendously compared to that of novel DPI formulations. It is not clear whether the device engineering alone would solve inhaled drug delivery problems. Therefore, a link needs to be established between developing smart formulation and or smart device, which ensure efficient and reproducible delivery of drug from the powder formulation. Pulmonary administration of medicaments is expanding with increasing rate of different diseases. In addition to asthma or COPD, some other DPI systems for mucolytics, antituberculosis, anticancer, antibiotics, drugs for sexual dysfunction, Augmentin® powder for otitis media, fentanyl for cancer pain, tobramycin (for cystic fibrosis long with infections related to chronic bronchitis and COPD), opioids for pain, interferons, alpha-1 antitrypsin, vaccines, gene therapy, and human growth hormone are in clinical development (Patton et al., 1999; Patton, 2005; Staniforth et al., 2006; Cheatham et al., 2006; Stephen and Babatunde, 2006; Chan et al., 2007). These studies reveal the promising future of DPIs in drug delivery and the application of DPI is expanding from pulmonary diseases to other disorders. Local and systemic delivery of different drugs for systemic chronic diseases needs to be focused more on using DPI formulations, which have a lot of potential. The DPI delivery systems are likely to contribute to successful drug delivery into the lungs not only to treat asthma, but also to deliver a wide range of therapeutic agents for pulmonary delivery. In future, very small amount of potent drugs like products of biotechnology will require smart devices that deliver drugs efficiently into the lower airway of lungs. Many devices mentioned in this review have yet to be commercialised; however, some of them will come to market in near future. Therefore, in combination with the increasing knowledge of DPI formulations and design of new devices, a step needs to be taken to develop more effective delivery system. The current trend in pulmonary drug delivery and potential benefits of this route, development of smart but reliable device will be continued to enhance deposition of drugs into deep lungs with a better patient compliance.
Designing of Azithromycine dry powder for pulmonary drug delivery:
The pulmonary delivery is a promising route for drug administration, both for effective local therapyand systemic administration. Aerosolized antibiotic agents have been used for the treatment of, and prophylaxis against, pulmonary infectionssince the 1950s mainly due to the factthat the targeted drug delivery can yield high concentrations at the site of infection in the lung while minimizing the systemic toxicity [1]. For the past few decades, the topical application of aerosolized antibiotics has been demonstrated for a number of compounds and indications, but more extensively for aminoglycosides in cystic fibrosis (CF), such as gentamicin [2,3] and the most frequently used tobramycin [4,5].
Azithromycin (AZI), which has been approved by the FDA for treatment of community acquired pneumonia and exacerbations of chronic obstructive pulmonary disease [6], was suggested by recent data [7] as the most promising anti-inflammatory therapy for patients with cystic fibrosis. Cystic fibrosis is a genetic disease that typically produces malnutrition and chronic respiratory infections, and ultimately leads to the death of most patients of this progressive respiratory disease [8]. In addition, several other respiratory disorders, like asthma, bronchiectasis, bronchiolitis obliterans syndrome after lung transplantation, diffuse panbronchiolitis, nontuberculous mycobacterial pulmonary diseases, pneumocystis jirovecii pneumonia and pulmonary nocardia infection, are attractive candidates for treatment with AZI [9]. AZI aerosols exhibit many advantages over the oral administration. Firstly, aerosolized AZI has a targeted effect without diffusing to other unnecessary sites in the body, consequently attaining higher concentrations in the lung (especially the epithelial lining fluid) [10], resulting in a dramatically reduced drug dose via pulmonary delivery. Secondly, oral AZI therapy is commonly associated with gastrointestinal symptoms and long-term use has been linked with hearing impairment, which can be avoided by the delivery of aerosolized AZI directly to the lung [6]. Finally, AZI, among all the macrolide antibiotics, has the strongest post-antibiotic effect (PAE) (up to 2.3-4.7h)[11],resultinginarelativelyshorttherapyperiod.Mostrecently, the firstaerosolizedAZI,the AZInebulizer[10],hasbeendevelopedfor the topical treatment of infections. To our knowledge, no DPI study on this drug has been reported yet, therefore, the final objective of this research is to develop a DPI of AZI as an alternative to the reported nebulizer.
Powder engineering, especially the selection of appropriate carriers as well as the control of formulation properties, plays a significant role in DPI investigation and a recent study even suggested the more critical role for powder formulation than the device design [12]. In this study, four candidate carriers were chosen: glycine, lactose, mannitol, and L-leucine. Glycien was chosen as it is a component of commercial Exubera®. The remaining candidates were used as they are recognized as safe and widely used carriers in previous DPI studies [13-18]. Spray drying, as a single-step processing technique with a great control over the particle characteristics [19-23] was applied for powder preparation. In-vitro deposition and main factors that have been reported to affect it [24], including morphology, powder density, hygroscopicity, flow ability, and particle size were all investigated in this study.
Materials:
AZI raw material (Batch no. 0504076) (Shanghai Modern Pudong Pharmaceutical Co., Ltd), lactose, phosphoric acid and sodium hydroxide (Tianjin Chemical Reagent Company), mannitol (Tianjin Damao Chemical Reagent Factory), L-leucine and glycine (Shanghai Kangda Amino acids Factory), potassium dihydrogen phosphate (Guangdong Shantou Xilong Chemical Factory), and ammonium phosphate mono- basic (Tianjin Bodi Chemical Co., Ltd) were obtained from the suppliers indicated.
Formulation design:
Dry powders for inhalation customarily consist of coarse carrier particles and micronized drugs and the dose of the active drug is often in a microgram amount. Inhalant powders in this study, however, were not produced into such an ‘ordered mixture’ but obtained by spray-drying a solution of drug and carrier. The reasoning behind this was based mainly on the relative large oral therapeutic dose of AZI (a single dose of 250 or 500 mg). A study on the effect of carrier type was carried out with four formulations, i.e., Lac-1, Man-1, Leu-1 and Gly-1 as listed in . The carrier:drug ratio was 1:5 for these formulations. To investigate the effect of carrier amount (drug load), three different carrier:drug ratios were applied with respect to each carrier. In particular, the carrier:drug ratios of sugar/polyol carriers were higher than those of amino acid carriers as the former is supposed to meet the need of extra bulking agents after the determination of exact drug dose via the pharmacodynamic effect study. In addition, a carrier-free formulation was also prepared as a control.
Preparation of AZI for pulmonary delivery:
A quantity of AZI raw material was weighed and added to a suitable volume of distilled water, and phosphoric acid was then added drop- wise with continuous stirring until a clear solution was obtained. The carrier, accurately weighed according to the carrier:drug ratios in , was dissolved in distilled water and then mixed with the drug solution. The pH of the obtained solution was adjusted with saturated sodium hydroxide solution to 7.0±0.1 which is reported to be suitable for the lung fluid environment [25], then diluted to volume and passed through a 0.22 mm cellulose acetate filter. The final solution was spray-dried using an SD-1000 spray dryer (EYELA, Japan) under the following conditions: temperature of the aqueous solution, ambient temperature; inlet temperature, 125±5 °C
Air flow rate, 0.60±0.05 m 3 min ; atomizing pressure, 15-17×10 kpa; pump rate level, 1.8-2.0 (approx. 6-7.5 ml min ). These conditions resulted in an outlet temperature of 75±5 °C. Once the aqueous solution was consumed, the outlet temperature was maintained at ~80 °C for approx. 15 min by regulating the inlet temperature so as to provide a secondary drying. The resulting dry powders were stored in a vacuum desiccator over silica gel until required for use. All the formulations were prepared from solutions with a same solid concentration (10%) following the same procedure and spray-drying conditions.
Characterization of spray-dried powders
a. Particle morphology observations :
A small amount of sample was scattered on mutually conductive double-sided adhesive tape placed on an aluminum stub and gold- coated using a JFC-1200 Fine Coater (JEOL, Japan) with a current of 20 mA for 200 s. Scanning electron micrographs were imaged with a scanning electron microscope (SEM) (SSX-550, Shimadzu, Japan) with an accelerating voltage of 15 kV and an emission current of 170 mA by scanning fields randomly at several suitable magnifications.
b. Powder density and flowability
The packed bulk density (r p) and the angle of repose as the most frequently used index for evaluating powder flowability were measured by a Powder Characteristics Tester PT-R (Hosokawa Micron Corp., Japan). The effective particle density (r e), which is necessary for the calculation of aerodynamic volume mean diameter (D a), could be obtained
c. Hygroscopicity test
Moisture is well known to affect powder cohesion through capillary force at high RH, therefore, a hygroscopicity test is critical for the optimization of powders for inhalation. Triplicate powders were precisely weighed for each formulation (m 1) and added to glass weighing bottles (50 mm×15 mm). All bottles were then placed in the simulated climate cabinet (CLIMACELL 222t, MMM Medcenter Einrichtungen GmbH, Germany) which was working fluently at the set temperature of 25±1 °C and at relative humidities (RH) of 30%, 45%, 60%, 75%, and 80%, respectively. After 24 h running of the unit, the bottles were taken out and weighed again (m 2). The %weight gained of each formulation after storing at each RH was then calculated from Eq. (2), and accordingly, the moisture sorption isotherms were made by plotting the %weight gained against the RH.
| Weight gained =(m2-m1)/m1 * 100%——————–2 |
The 60% RH was used to examine the moisture absorption rate because this RH value proved to be the identical inflection point in all the moisture sorption isotherms and rather, at 60% RH, powders containing sugars/polyols dissolved in the sorbed moisture after 24 h. The %weight gained was measured after 2, 4, 8, 12, 16, 20 and 24 h storing. A linear equation was made by plotting %weight gained against the corresponding time and the slope was considered as the moisture absorption rate (MAR).
d. Particle size analysis
The particle size was determined by both laser diffraction and Time-of-flight (TOF) techniques in order to obtain more reliable results. A Beckman Coulter LS 230 particle size analyzer was applied for particle sizing by laser diffraction. In this method, powders were poured carefully into the sample cup until the obscuration reported on the title bar of the run window reached about 4%. The measurement was then performed for a sample run time of 90 s. Each sample was analyzed in triplicate. The theoretical estimate of the aerodynamic volume mean diameter (D a) can be obtained, according to Eq. (3) [27], from the geometrical volume mean diameter (D g), which can be read directly in the analysis report. The data obtained were also expressed in terms of the particle diameter at 10% (D 10), 50% (D 50) and 90% (D 90) of the volume distribution. Thus, the span, as an index of the width of the distribution relative to the median diameter D 50, was obtained from Eq. (4) [27].
A particle size measurement based on the TOF technique was obtained using the PSD 3603 particle size distribution analyzer (TSI inc., USA). Both the geometrical and the aerodynamic volume mean diameters were measured after typing in the effective particle density of powders in question. Apart from the particle size, the specific surface area (SSA) calculated based on Eq. given bellow was also presented in the statistical table. Each sample was analyzed at least three times.
Influence of carrier types on AZI powders for inhalation:
a. Influence of carrier types on powder morphology, density and hygroscopicity:
It was interesting to find in this study that the type of carrier profoundly influenced the morphology of spray-dried AZI powders. As shown in Fig. 2, the carrier-free formulation (Fig. 2a) and amino acid based formulations (Fig. 2d–e) showed a similar dice-like appearance. It is notable that scanning electron micrograph of powders containing L-leucine (Fig. 2d) displayed the most uniform distribution, which was in good agreement with the particle size distribution results as shown in Fig. 3a. The addition of lactose (Fig. 2b) resulted in wrinkled particles, which were similar to the appearance of spray-dried bovine serum albumin (BSA) described by Chew et al [29]. Unlike lactose, mannitol produced more globular and smooth particles (Fig. 2c). Indeed, it is difficult to distinguish the exact reasons for such differences in morphology. However, it is supposed that the significant difference between lactose and mannitol based formulations could be partially caused by the fact that the drying process often occurs from the surface part of the liquid mannitol droplet while the aqueous solution of lactose can condense into a high concentration instead of recrystallization, which is basically because mannitol recrystallizes easily with a much lower glass transition temperature (Tg) of 4 °C [30] compared to lactose with a Tg of 108 °C [31]. As listed in Table 2, the increasing order of powder density with four carrier based formulations which had the same carrier:drug ratio of 1:5 is Gly-1bMan-1bLeu-1bLac-1. Unfortunately, it was indicated that none of these four carriers had significant effects on decreasing the density of spray-dried AZI powders, even for L-leucine which was ever reported as an ultra low density additive [14]. Moreover, densities obtained in this study were higher than most of the reported values of other DPI products. This unexpected finding is probably owing to the nature of the model drug AZI as well as the nonporous solid morphology of the formulations.
- b. Influence of carrier amount on AZI powders for inhalation :
The carrier amount showed no significant influence on the powder morphology (Figures not shown), density, and hygroscopi- city (Table 3). Added to this, one could draw a conclusion through the earlier study on the effect of carrier type that powder flow ability and particle size are two dominant factors determining the aerosolization performance of DPI containing AZI. Hence, these two important properties, along with the in-vitro deposition behavior were discussed in more detail in this part in terms of the effect of carrier amount.
- c. Influence of carrier amount on powder flowability
Spray-dried powders of pure AZI showed a poor flowability and easily adhered to the walls of the container. In terms of lactose and glycine, carrier amount had no obvious influence on the flowability as no obvious change in the angle of repose was detected with the variation in their amount as listed in However, the addition of the other two carriers, i.e. mannitol and L-leucine, in proportion to the amount, properly improved the powder flowability and hence made the handling much easier. More specifically, as the L-leucine: AZI ratio approached 1:5, the angle of repose decreased significantly from 62°(carrier-free) to 43°, indicating a great increase in flowability. This increase could be partially attributed to the decrease in SSA from 2.79 m 2 g (the carrier-free formulation) to 1.96 m 2 g . The reason for the change of flowability with the variation in the amount of mannitol is that spray-dried mannitol was reported to be character- ized with 100% crystalline [35], and furthermore, it was reported that powders with higher crystalline content had lower surface energy, less cohesion and concomitantly better flowability [36,37]
d. Influence of carrier amount on particle size:
As the amount of mannitol increased, the D g value increased from 4.35 mm by laser light diffraction and 6.82 mm by TOF technique to 10.85 mm and 13.71 mm, respectively. However, changes in lactose amount had no significant effect on particle size, which could be well reflected by the horizontal line formed with three points of formulations Lac-1, Lac-2 and Lac-3 in Fig. 4. The relationship between particle size and the amount of amino acids was not linear. Interestingly, no significant difference in particle size was found between formulations with low and moderate amounts of L-leucine, however, the particle size with the high amount increased to 4.31 mm by the laser light diffraction technique and 6.74 mm by the TOF technique. On the contrary, glycine in moderate and high amounts showed similar small particle sizes while relativelylarge particles were produced when a low amount was added. These findings are likely to be linked to certain reactions between drug and carrier, which are particularly dependent on the amount of carrier. However, this is just a supposition and real reasons for such findings still need to be further explored. In addition, as displayed in Fig. 4, it was reproduced in this study that large particles correspond to small SSA in terms of particles in similar shapes.
6. A new approach for treating Broncho-constrictive condition using nano-salbutamol Dry powder for pulmonary drag delivery:–
Introduction:
Worldwide, more than 300 million people suffer from asthma or COPD [1,2]. It is well known that the local drug delivery of beta-agonists to the respiratory tract offers several advantages over the systemic route in treating broncho-constrictive conditions [3–7]. Compressed air nebulization, metered dose inhalation and dry powder inhalation are now commonly used for targeting drug delivery to the tracheo-bronchial region. However there are several features associated with inhalation therapy that requires improvement. Firstly, deposition in the target area is just 5–15% [8] that reduces further in patients with dyspnoea or mucus plugs in the respiratory pathway. Pharmacological effect of the inhaled drug tends to be short, mostly not more than 1–2 h [9]. In pediatric age group asthma and in advanced COPD small airway involvement are predominant, and inhalation drug delivery systems are not much effective because of poor reach and deposition in small airways. A major portion of the inhaled drug is predominantly swallowed into the esophagus that may lead to acid-induced esophagitis besides drug wastage. Within the inhalation technologies, DPI is favored because of simplicity, ease of use and costeffectivity. Salbutamol sulphate dry powder inhalation (SBS DPI) is the most important member of this family. Drug particles of SBS in the size range of 5–15 lm have been found therapeutically important because it is this respiratory fraction that coincides with deposition in the target area of trachiobronchial region. Role of micronized SBS DPI is, however, limited to prophylaxis and is used in mild cases. It is not considered potent enough for the treatment of moderate or severe cases [10]. Bronchodilation may be suboptimal or even non-existing because of proximal deposition of drug particles in the presence of mucus, inflammatory fluid, tortuous respiratory pathway and reduced lumen, and poor inspiratory capacity in chronic or severe cases [9]. Clearly, more research isrequired to make SBS DPI more effective in conditions where it presently has a limited role as a preventive or an adjunct. It is known that the deposition of inhaled aerosol is related to the particle size; smaller particles tend to travel farther and settle in the deeper and smaller airways [11]. Submicron-sized particles tend to deposit in small bronchi and bronchioles while nanoparticles reach the more peripheral portions of the respiratory tree and alveoli. Such particles are able to negotiate tortuous pathways and mucus plugs better. In the management of asthma and COPD, one of the recent strategies has been the reduction of drug particles size to 1–5 lm level to enhance penetrability, particularly in the presence of inflammatory, obstructive or constrictive factors so that the drug could be directed to the target area in pharmacological dose [12,13]. Several authors have recently demonstrated the changed deposition pattern of the smaller particles using scintigraphy [14]. However, there appears to be a limit for reducing the particle size for enhancing penetration because particles with even lesser size (less than 1 lm, and particularly nanoparticles, less than 400 nm) may not be suitable as delivery system due to mismatched targeting and the fact that inhaled nanoparticles tend to be exhaled significantly [15]. Nanoparticles are solid colloidal particles ranging in size from 10 to 1000 nm and hold a promising role for the respiratory drug delivery [16]. Both in vitro and in vivo studies have demonstrated that nanoparticles are promising carrier systems for respiratory drug targeting, particularly to the alveoli [17–19]. However, their clinical role in respiratory tract diseases remains unclear. In bronchial asthma and COPD particularly, many researchers feel that nanoparticles may not have a role because of altogether different targeting. We aimed to improve the efficacy of SBS DPI in the management of bronchial asthma and COPD using inhalation therapy for alveolar deposition (ITAD) concept. Our concept is based on the premise that although submicron-sized particles of SBS may not be targeted to the sites involved in bronchial asthma and COPD, yet these may possess certain advantages not associated with micro- SBS DPI. Our work is based on three hypotheses. Micron-sized colloidal particles readily undergo phagocytosis by the macrophages stationed in the respiratory tract while nanoparticles are largely spared [20–22].This will lead to entrapment and reduction in pharmacological action of micronized drug despite being present in pharmacological quantity. The macrophages might release soluble SBS later on but the concentration by then will be too low for clinical benefit. Patients with asthma and COPD have several folds higher number of macrophages due to inflammation. Our second hypothesis was that the distally deposited nano-SBS particles will move proximally by mucociliary action and will contribute to pharmacological action at the target site. Our third hypothesis was that nano-SBS may enhance the fraction of inhaled SBS deposited in the target spaces (10–15% dose). Significantly, 60–70% of micronized SBS DPI is trapped in the pharynx, adsorbed to the much larger lactose particle. If nano-SBS could be shown to have a better respirable fraction, this could lead to more suitable pharmacokinetics
Materials and method:
Micronised SBS (commercial) was supplied by Cipla Ltd. (Mumbai, India). Lactose (Pharmatose_) was obtained from DMV international (Veghal, The Netherlands). HPLC grade acetonitrile was purchased from Merck (India). Water used was purified by reverse osmosis (MilliQ, Millipore, USA). All others chemicals were of analytical grade.
Characterization of nano-SBS:
Scanning electron microscopy (SEM)
SEM photographs of nano-SBS were taken by scanning electron microscope (Leo 435VP, Cambridge, UK). The samples were mounted on an aluminium stub and coated with gold in an argon atmosphere at 50 mA for 100 s using balzers SCD020 sputter coater unit (BAL-TEC GmbH, Witten, Germany).
Quasi-elastic light scattering (QELS)
The particle size distribution for the nanoparticles was analyzed by quasi-elastic light scattering (QELS). The prepared nanosuspension was taken in a test tube and placed in the goniometer and was analyzed by QELS, the results were recorded on the histograms.
Fourier transforms infrared spectrometry (FT-IR)
FT-IR spectra were recorded with a FT-IR JASCO, Easton, MD spectrometer in range 400–4000 cm_1 using a resolution of 4 cm_1 and 10 scans, to evaluate the molecular states of micronized SBS and nano-SBS. Samples were mixed with potassium bromide (KBr) and were pressed to obtain supporting disks.
Differential scanning calorimetry (DSC)
The phase transition of micronized and nano-SBS was analyzed
by differential scanning calorimeter (Pyris 6, Perkin-Elmer, USA). The DSC analysis was conducted over a heating range of 50–250 _C under controlled conditions. 2.3.5. X-ray diffraction (XRD) The physical state of nano-SBS was studied by means of XRD patterns and compared to micronized SBS. Phase identification was conducted using an X-ray powder diffractometer (Shimadzu XRD-6000, Mumbai, India) with Cu Ka radiation at a scanning speed of 0.05_/min.
Characterization of nano-SBM DPI Formulation:
The aerodynamic particle deposition of SBS DPI was measured using an Anderson Cascade Impactor (ACI) (Copley Scientific, Notttingham,UK). The ACI consisted of initiation port (IP) and the preseparator (PS), seven stages and a final collection filter. The stages were coated with polypropylene glycol dissolved in hexane (2% w/w). The stages were left to dry under ambient room conditions for 1 h. Experiments were conducted at an air flow rate of 60 L min_1 for 4 s. The operating conditions and theoretical cutoff
Time per actuation (s)
Volume per actuation (L)
Cut-off diameter (lm)
Operating conditions and theoretical cut-off diameters of Anderson cascade impactor.
Anderson cascade impactor
diameters are shown in Table 1. After actuation, the wash solutions from different parts were collected and quantitated for drug content by UV spectrophotometer. Respirable particle fraction (RF) and emission dose (ED) were calculated to describe the inhalation properties of DPIs. The measurements were performed in six replicates
Ventilation scintigraphy with Tc-99m nano-SBM DPI:
Ten healthy volunteers were recruited for ventilation scintigraphy study with Tc-99m nano-salbutamol DPI (7 males, 3 females, mean age 35 years, 22–54 years). The clinical protocol was approved by the institutional human Ethics Committee (Reg. No.INM/TS/IEC/006-017/07). Prior to dosing, subjects were trained to inhale dummy DPIs deeply and to retain the breath following deep inspiration for 10 s. The volunteers were trained to exhale into an 8-L polythene bag to collect the air expired out. Vital signs were recorded before and 30 min after each dose and before discharging the subject from the study center. Adverse events were monitored throughout the study. The human studies were conducted on a dual head Hawkeye gamma camera system. A single gelatin capsule containing freshly prepared 1.5–2 MBq Tc-99m nano-SBM (and 200 mcg of SBM) was given to each volunteer after obtaining the radioactivity count rate of the capsule on the Gamma Camera. Upon inhalation, twodimensional scintigraphic image of the chest region was obtained for 300 s covering the highest and lowest point of radioactivity distribution (oropharynx to stomach). It was followed by a 5-min image of the polybag containing the exhaled air. Count rate of the used capsule and the Rotacap device was found. All images were recorded on a computer system assisted with the software Entegra Version-2. Additional images of the chest were taken between 2 and 4 h. This was done to observe the movement of radioactivity if any. Krypton-81 ventilation scan was not performed because the lung images generated by inhaled radiolabeled nanoparticles showed the lung contours prominently. Also, the main purpose of this study was to test whether the nanoparticles shifted proximally from lung spaces in time or not, which did not specifically required this additional scan.
Scan analysis:
Images acquired on the same time scale ensured that the count statistics comparisons between different scans were valid. Region of Interest software was drawn around the oropharynx, esophagus, stomach, and whole lung for obtaining count statistics. The lung region was again subdivided into central, intermediate and peripheral sections which translate predominantly into respiratory tree, mixed and alveolar region, respectively [26]. The peripheral lung zone to central lung zone deposition ratio (P/C ratio) was calculated as an index of regional lung deposition. Because swallowing action continuously transports radioactivity deposited in oropharynx into stomach, counts deposited in these two organs were integrated to represent a single compartment. Respiratory fraction, the fractions of radio labeled drug deposited in the central, intermediate and peripheral lung, was calculated in the initial and subsequent images. Visual comparison between the lung images was done to record movement of the deposited drug with time from one compartment to another.
Quality analysis of Tc-99m salbutamol sulphate nanoparticles:
Radiolabeling stability. The radiolabeling efficiency at the time of complexation was 92–96% while it was 89–92% after 6 h of dry incubation. This is considered adequate for in vivo imaging by scintigraphy norms. Using different solvents, the amount of reduced hydrolyzed technetium was found to be 2–5%
Preparation of Poly (ether-anhydride) dry powder aerosols for sustained pulmonary drug delivery:–
A new family of high molecular weight polymers designed for controlled drug delivery following inhalation or injection, the poly(etheranhydrides), has been developed recently [8]. These materials show promise for use in the lungs for several reasons: (i) they are composed of chemical species that are known to be safe in humans for various applications, poly(ethylene glycol) (PEG), sebacic acid (SA) and 1,3-bis(carboxyphenoxy)propane (CPP); (ii) their erosion times can be controlled over periods ranging from hours to several weeks by varying their monomer composition, thereby regulating the release of entrapped therapeutics; (iii) they deliver drugs with molecular weights ranging from 443 to over 5_106 Da in a continuous fashion for up to 7 days [9], whereas commonly used PLGA polymers often release drugs in a triphasic manner [10]; and (iv) the addition of PEG into the polymer backbone can decrease particle clearance rates by phagocytosis in the deep lung. However, effective aerosolization of particles made using the ether-anhydride family of polymers is necessary to realize their potential benefits for controlled drug delivery via the lungs. Aerosolization and lung deposition of microparticles are significantly affected by particle physical and chemical properties [11]. The key physical parameter that predicts the site of aerosol deposition within the lungs for particles larger than several hundred nanometers is the aerodynamic diameter (daero) [12]. Even with an optimized aerodynamic diameter, however, particle surface properties can cause significant aggregation of micron-sized particles, reducing the efficiency of deep lung deposition. Surface properties, such as hydrophilicity, rugosity and charge, control interparticle adhesion forces that influence dry powder aerosolization from an inhaler and, subsequently, deposition in the lungs
Material:
Sebacic anhydride (acyl-SA) and polyethylene glycol anhydride (acyl-PEG8000) were synthesized as previously described [8]. PEG had a molecular weight of 8000 Da prior to acylation. Polyvinyl alcohol (PVA; 88 mol% hydrolyzed, Mw = 78 kDa, Polysciences, Warrington, PA), bovine serum albumin (BSA) (Sigma, St. Louis, MO), FITC-Dextran (Mw = 20 kDa, Sigma), lactose (Sigma) and other reagents were used as received without further purification.
Polymer characteristics:
Synthesis and weight analysis:
Acyl-PEG and acyl-SA powders were mixed in defined ratios in round-bottom flasks equipped with a stopcock adapter and polymerized by melt polycondensation at 180jC under high vacuum for 30 min. Briefly, the flask was immersed in an oil bath maintained at 180 jC and the monomers were allowed to melt. High vacuum was applied (f0.04–0.05 torr) and the condensation byproduct, acetic anhydride, was collected in a liquid nitrogen trap. The polymers were allowed to cool completely at the end of the reaction and they were then dissolved in chloroform. The solution was precipitated dropwise into excess petroleum ether. The precipitate was collected by filtration and dried under vacuum to constant weight. The molecular weight and polydispersity of the polymers were determined by gel permeation chromatography (GPC) using a JASCO PU-980 intelligent HPLC pump, 1560 intelligent column thermoset and RI-1530 intelligent refractive index detector (JASCO, Easton, MD). Samples were filtered and eluted in chloroform through a series of Styragel columns (guard, HR4 and HR3 Waters Styragel columns) at a flow rate of 0.3 ml/min. Weight– and number–average molecular weights were determined relative to polystyrene standards (Fluka, Milwaukee, WI).
Polymer contact angle measurements:
The relative polymer hydrophobicity was determined by contact angle measurements (Reme´-Hart Contact Angle Goniometer, Mountain Lakes, NJ). Uniform polymer surfaces were obtained by film casting a solution of polymer dissolved in chloroform onto glass slides, followed by evaporation of the chloroform. A 20-Al drop of water was pipette onto the polymer surface and the contact angle measured. Reported values represent the average of 4–10 samples.
Imaging of micro-particle:
Differential interference contrast (DIC) and fluorescence images of drug carriers were acquired with alaser scanning confocal microscope (Zeiss LSM 510 Meta, New York, NY) equipped with a 100_ oil immersion lens and a 488-nm laser line. Samples were prepared by suspending 5–10 mg of particles in 1 ml water, then placing the particle suspension between a glass slide and coverslip. Aerosol particle surface morphology was evaluated by scanning electron microscopy (SEM) with a Jeol JSM-6700F cold cathode field emission SEM (Peabody, MA). Microparticle samples were attached to SEM mounts using double-sided graphite carbon tape and sputter coated with platinum at 0.5 nm/min for 5 min using a Polaron Range SC7640 Sputter Coater (Quorum Technologies, Newhaven, England). Populations representative of each microsphere sample were photographed.
Micro-particle bulk characterization:
The volume–mean size distribution and geometric standard deviation (GSD) of microparticles were determined using a Coulter Multisizer IIe (Beckman- Coulter Fullerton, CA). To allow calculation of the theoretical particle mass–mean aerodynamic diameter (see Eq. (2) in Section 3), all particles within a given formulation were assumed to possess roughly the same density. Therefore, the volume– mean particle diameter was approximated as roughly equal to the mass–mean diameter. Approximately 2 ml of isoton II solution was added to 5–10 mg microparticles. The solution was briefly vortexed to suspend the microparticles and added drop wise to 100 ml of isoton II solution until the coincidence of particles was between 8% and 10%. Greater than 100,000 particles were sized for each batch of microparticles to determine the mean particle size and size distribution. The bulk density of the particles was determined by tap density measurements. Briefly, particles were loaded into 0.3-ml sections of a 1-ml plastic pipette, capped with NMR tube caps and tapped approximately 300–500 times on a hard benchtop until the volume of the powder did not change. The tap density was determined from the difference between the weight of the pipette before and after loading, divided by the volume of powder after tapping. Thermo gravimetric analysis was conducted to determine particle water content using a TA Instruments SDT 2960 apparatus (10 C/min, 25–250 C; New Castle, DE)
on 6–12-mg samples under nitrogen purge
Effect of aerodynamic size on
particle:
To determine the effect of poly(ether-anhydride) particle aerodynamic size on aerosolization and deposition, poly(PEG:SA//10:90) particles with aerodynamic diameters of 2.7, 3.4 and 4.0 Am were produced (Table 3). Microparticle size had no significant effect on the ED of poly(PEG:SA//10:90) particles in the aerodynamic size range of 2.7–4.0 Am (Fig. 2A). In addition, a change in aerodynamic diameter from 4.0 to 3.4 Am caused no statistically significant change in the fine particle fraction in the in vitro lung model (Fig. 2B). However, a decrease in aerodynamic diameter to 2.7 Am nearly doubled the FPF, from 16% to 30% ( p < 0.02 compared to either 3.4 or 4.0 Am particles) (Fig. 2B), as demonstrated by a shift in the deposition pattern towards the lower stages of the impactor (stages 3–7) (Fig. 2C).
Particle shape and surface roughness:
Confocal and light microscopy images (Fig. 3) show a significant difference in internal structure between the poly(SA) microparticles and poly (PEG:SA) microparticles. Poly(SA) particles have smooth surfaces and a thin outer polymer shell, with the model drug FITC-dextran evenly distributed within the inner core of the particles. On the other hand, poly(PEG:SA//10:90) microparticles have a globular internal structure, with the drug distributed in small pockets throughout the particle (presumably corresponding to the water phase of the primary emulsion). Poly(PEG:SA//30:70) microparticles exhibited a mix of the two types of internal structures. SEM showed that all formulations produced mainly spherical particles (Fig. 4). Poly(SA) microparticles had slightly textured, but relatively smooth surfaces (Fig. 4A). Polymers containing 10% PEG produced particles with rougher surfaces and holes that penetrated into the particle (Fig. 4B). Particles made with polymers containing 30% PEG appeared smoother than the 10% PEG polymer particles, but had noticeably larger holes (Fig. 4C). SEM on a mixture of lactose particles and polymer microparticles showed very large, blocky lactose particles with many smaller microparticles attached, typically at the edges and crevices (Fig. 4D). Particles were rarely attached to the flat planes of the lactose particles unless smaller lactose particles were present on those surfaces.
Fig. 3. Confocal images overlaid with light microscopy images showing the internal texture of polymeric microparticles and the location of FITC-dextran encapsulated within the particles: (A) poly(SA), (B) poly(PEG:SA//10:90), (C) poly(PEG:SA//30:70) microparticles.
Conclusions
Biodegradable poly(ether-anhydride) microparticles have been designed for drug delivery following aerosolization into the lungs. PEG added to the polymer backbone significantly changed particle hydrophilicity, surface roughness, surface charge and water content and may provide a steric barrier to reduce particle aggregation.
Pulmonary delivery of therapeutic peptides via dry powder inhalation–
General Introduction :
Preparing dry powder formulations for inhalation is an attractive and appreciated proposition because many solubility and stability issues can be avoided [7,8]. Furthermore, a dry powder aerosol offers the capacity to provide a wide range of single doses per inhalation. Further advantages are the low susceptibility to microbial growth and the suitability for both water soluble and insoluble drugs [9]. However, the small particle size necessary to achieve effective lung deposition causes problems in processing (poor flowability) and redispersion (strong agglomeration and adhesion) [10,11]. Protein therapeutics require an accurate dose consistency, physico-chemical stability as well as a constant and efficient deposition in the respiratory tract [12]. Moreover, some may require the administration of relatively high doses. The production of a high-efficiency inhalation device therefore is a major issue. All passive dry powder inhalers rely on the inspiratory flow from the patient’s inhalation. The majority of drugs in powders for inhalation is prepared by jet milling which can also be used for the preparation of fine particles of protein and peptide molecules [7,14,15]. Spray drying has been recognised as a successful process to generate protein-containing powders in a single step from solutions [16]. Within the spray drying process, an aerosol is usually generated by pneumatic nozzles. There are some distinct disadvantages associated with these nozzle systems, such as a control over the mean droplet size, a broad droplet distribution, and the risk of clogging in the case of suspensions. Ultrasonic nozzles are able to generate droplets with a more uniform size which could lead to a relatively homogeneous size distribution of the produced powders [17]. Using such powders may result in a more accurate delivery to the airways [18]. Supercritical fluid technology [19] and spray freeze drying [20] are also promising techniques for producing small particles of peptides and proteins. A method of liquid phase milling in fluid propellant for the preparation of peptide suspensions for pressurised metered dose inhalers has been recommended by Adjei [21]. Lizio [22] improved this technique using a modified and abrasive resistant pearl mill. Fine protein particles with a volumetric mean diameter of 3.1 mm could be produced without the generation of degradation products or contaminants deriving from the milling process. The system also allowed a direct manufacturing of fine drug suspensions for systemic aerosol delivery by pressurised metered dose inhalers [23].
Materials and analysis :
Cetrorelix-acetate was synthesised for preclinical pharmacokinetic studies in the laboratories of Degussa AG (Hanau, Germany). Crystalline alpha lactose monohydrate (CapsuLacw 60) was fabricated by Meggle GmbH (Wasserburg, Germany). The pearl mill micronisation was performed in Solkanew 227 (Heptafluorpropane, HFA 227, Solvay Fluor und Derivate GmbH, Hannover, Germany). Anhydrous ethanol and ethanol 96% were applied during pearl mill micronisation and supplied by Merck Eurolab GmbH (Darmstadt, Germany). Acetonitrile and trifluoracetic acid were used for HPLC analyses and supplied by Merck Eurolab GmbH. All quantitative analyses of cetrorelix-acetate were performed by reversed phase HPLC provided with a Hewlett Packard 1100 evaluation unit (Agilent Technologies Inc., Avondale, USA). The separation column was purchased from Macherey-Nagel GmbH (Du¨ren, Germany). Details are summarised in Table 1. Also, potential degradation products resulting from the micronisation processes have been considered by HPLC
Particle size distribution:
The particle size distribution of the micronised cetrorelix batches was determined by laser light scattering. Using a wet dispersion method the analyses were performed on a Master Sizer X (Malvern Instruments GmbH, Herrenberg, Germany), equipped with a 45 mm focus lens which covers a particle size range of 0.1–80 mm. Small amounts of powder were suspended in a mixture of cyclohexane and Spanw 85. In order to deaggregate particles, the samples were treated for a short time in an ultrasonic bath before measurement.
Each sample was assayed in triplicate. The standard algorithm of Malvern software based on the Mie theory was used for calculating the volumetric particle size distribution. The key parameters D10, D50 (mediandiameter) and D90 represent the volume diameters at the 10th, 50th and 90th percentiles of this distribution.
Determination of water content:
The water content of three selected formulations was analysed according to the Karl Fischer moisture method of the European Pharmacopoeia (method A) [29]. The measurements were performed with a Titrinow 701 KF (Metrohm Ltd, Herisau, Switzerland).
Micronisation methods:
Micronisation by spray drying
The spray drying of cetrorelix-acetate was targeted at producing a narrow particle size range around 3.0 mm. A pilot scale spray dryer was used, constructed and operated by Degussa AG (Hanau, Germany). The spray generator at the top of the spray tower was composed of an atomiser nozzle and the required control logic. In order to control the process by optical criterion, the atomisation process was watched by a CCD-
camera. The air passage was optimally adapted by a novel technology aimed at the production of mono sized droplets caused by Rayleigh jet breakup conditions (filed for patent). Protein aceous solutions of 0.016% (w/w) of cetrorelix-acetate were prepared in acetic acid (94% w/w) and continuously fed to the spray dryer at a solution flow rate of 1.35–1.55 kg/h and an aspiration of 30–33 Nl/h. During spray drying an inlet temperature of 155–165 8C and a spray drying an inlet temperature of 155–165 8C and a product temperature of 49–57 8C were kept
Injection volume (ml)
Flow rate (ml/min)
Oven temperature (8C)
Detection wavelength (nm)
Attenuation (mAU)
Acetonitrile (ml)
Trifluoracetic acid (ml)
Eluent A: (58%)
Eluent B: (42%
Micronisation by milling:
A pearl mill (Dispermatw SL-C 12, VMA-Getzmann GmbH, Reichshof, Germany) was specially modified for the present study: Its milling chamber was provided with iridium-stabilised ZrO2 as abrasion-resistant material. This was also used for coating the rotating pearls (diameter of 0.6 mm). Further on, special linking tubes for in-process testing and sample collection were positioned on the reflux tube. A cryostat (N8-KT90W, Gebru¨der HAAKE GmbH, Karlsruhe, Germany) for temperatures down to 270 8C was coupled with the pearl mill and ethanol 96% was used as coolant. At the beginning of the micronisation process, cetrorelix-acetate was suspended within fluid propellant (Solkanew 227) using the share forces of an UltraTurraxw at 8000 rpm (IKA Werke, Janke & Kunkel GmbH, Staufen, Germany). The resulting suspension was filled into the modified Dispermatw. The milling process was performed according to the operative parameters described elsewhere [24]. The suspension of milled cetrorelix-acetate in Solkanew 227 was finally filled into a flask and rotated at room temperature until the fluid propellant was completely evaporated.
Preparation of adhesive mixtures:
A free flowing and coarse lactose (CapsuLacw 60) was added as a carrier over which the drug particles could be distributed. The carrier and also the preparation process of the drug mixture were chosen
to achieve a homogeneous distribution of the active
ingredient within the powder blend. The bulk homogeneity of each mixture was tested by taking 10 random samples (mass corresponding to the device metered mass). The drug content was determined by HPLC as described above. Afterwards, the blends were filled into the powder cartridges of the dry powder inhaler described below. Further on, sufficient flow properties for an accurate dose metering were targeted, as well as an effective de-agglomeration of the powder mixture
into a mono-particulate aerosol. Table 2 gives an
overview of the investigated batches which were
manufactured by two different preparation methods.
Formulation Method- A and B:
Overview of the prepared adhesive mixtures with CapsuLac w 60 as carrier
and different drug loads of pearl milled and spray dried cetrorelix-acetate,
respectively
Preparation method:
A (dry mixing procedure) Micronised cetrorelix-acetate was added to the lactosecarrier and initially blended with a metallic spoon to breakup large agglomerates. After passing through a 250 mm stainless steel sieve, the mixture was blended for 60 min at 40–50 rpm using a free fall mixer. Powder blends of four different mass fractions of cetrorelix-acetate (5–20%) were prepared in batches of 20 g with pearl milled drug (batches PM-20, PM-15, PM-10, PM-5) as well as spray dried drug (batches SD-20, SD-15, SD-10, SD-5).
Method B (wet mixing procedure) A suspension of cetrorelix-acetate in fluid propellant was prepared using the share forces of an UltraTurraxw for 2 min at 8000 rpm (IKA Werke, Janke and Kunkel GmbH, Staufen, Germany). The mixture was afterwards filled into a flask which contained lactose also suspended in fluid propellant. The lactose-cetrorelix suspension was rotated at room temperature (70–100 rpm) until the fluid propellant was completely evaporated. The resulting dry powder contained 11.5% cetrorelix-acetate (batch PMS-10).
Dry powder inhaler:
All studies on inhalation performance were carried out with the Novolizerw (Sofotec GmbH, Frankfurt, Germany). Fig. 1 presents the main components of the device. The inhaler’s cartridges were filled with the powder blends of lactose and micronised cetrorelix and placed in the device. During each actuation, the metering cavity of a cartridge was filled with formulation from the powder reservoir. A reproducible dosing was achieved through an optimal interaction between the device metering system and the powder formulation’s rheological properties. In our study the metering performance of each formulation was investigated by weighing 20 doses metered by actuation. The drug dose fell into the powder channel when released from the metering cavity. During inhalation the cyclone-based circulation chamber caused a de-agglomeration of drug and carrier. Ideally an aerosol cloud of non agglomerated single drug particles left the inhaler.
Fig. 1. Schematic design of the dry powder inhaler (Novolizerw) used within the study. The metering cavity of the cartridge is filled with formulation from the powder reservoir. A cyclone-based circulation chamber causes the de-agglomeration of drug and carrier.
Conclusions
Pearl milling was an effective but mild micronisation technique for the decapeptide cetrorelix. Formulated as highly loaded adhesive mixture, high fine particle fractions could be generated with the de-agglomeration system of a multidose dry powder inhaler (Novolizerw). The best performance was shown by a formulation manufactured via wet mixing of a drug-carrier-suspension (formulation PMS-10). Fine particle fractions of about 60% wereobtained. Within the range of airflow rates typically generated in vivo through the Novolizerw, no significant effect on the dispersion and the resulting amount of respirable drug was determined
Development of Dry powder system of novel Vasoactive intestinal peptide analog for pulmonary drag delivery:
General information about VIP:
Vasoactive intestinal peptide (VIP), consisting of 28-amino acid peptide, is widely distributed in the central and peripheral nervous systems (Fahrenkrug, 1979). VIP is abundantly present in normal human lung (Paul and Said, 1987) and VIPimmunoreactive nerves are present in the smooth muscle layer and glands of airways, and within the walls of pulmonary and bronchial vessels (Dey et al., 1981; Laitinen et al., 1985). VIP exerts diverse biological effects, such as smooth muscle relaxation, suppression of inflammation, neuromodulation and immunomodulation (Said, 1982; Bellinger et al., 1996; Gomariz et al., 2001), which are mediated through interaction with two receptor subtypes named VPAC1 and VPAC2 receptor (Harmar et al., 1998). These G-protein coupled receptors were cloned and identified in the lung of several species (Lamet al., 1990; Ishihara et al., 1992; Sreedharan et al., 1993). VIP has been shown to act as a potent bronchodilator and the deficiency of VIP in the airways was suggested as a possible pathogenic factor in asthma (Ollerenshaw et al., 1989). It has been also suggested that VIP enhances synthesis of phosphatidylcholine, the major component of pulmonary surfactants, by enhancing choline-phosphate cytidylyltransferase (CCT) and CCTα mRNA level via VIP receptor-mediated pathway (Li et al., 2004).Based upon these findings, administration of exogenous VIP may be effective for the clinical treatment of asthma. However, VIP is susceptible to the rapid chemical and biochemical digestion after systemic administration, especially the enzymatic digestion induced by neutral endopeptidase, mast cell tryptase and chymase (Caughey et al., 1988; Goetzl et al., 1989; Tam and Caughey, 1990) could be significant. Herein, the rapid degradation of VIP after the systemic administration is the part of reason for the limitation of its clinical applications. Therefore, for the clinical application of VIP as a therapeutic agent, a metabolically stable analogue of VIP needs to be developed. In addition to the stability issue, there is the probability that systemic administration of VIP causes cardiovascular side effect (Morice et al., 1983), therefore the effective drug delivery system also should be applied for the specific delivery of VIP to the target tissue. Actually, the development of several drug delivery system for VIP has been attempted and they include nasal route (Dufes et al., 2003) and vector-mediated delivery (Bickel et al., 1993). Recently we have synthesized a novel derivative of VIP [R15, 20, 21, L17]-VIP-GRR (IK312532), which was designed to enhance its stability against the enzymatic digestion (Onoue et al., 2004). IK312532 was found to be a potent VPAC receptor agonist and it significantly relaxed tracheal smooth muscle in a dose-dependent manner (Ohmori et al., 2004). These findings urged us to develop the formulation for pulmonary administration of novel VIP analogue.
Materials:
VIP and [R15, 20, 21, L17]-VIP-GRR(IK312532) (Table 1)were synthesized by a solid-phase strategy employing optimal protection as reported previously (Merrifield, 1969). [125I]VIP (81.4 TBq/ mmol) was purchased from DuPont-NEM (Boston, MA). All other chemicals were obtained from commercial sources.
Preparation of dry powder formulations:
Dry powder formulation of peptides was prepared as reported previously (Endo et al., 2005). Briefly, peptides and excipients were first ground to fine powders with a pestle and mortar and then milled with an A-O JET MILL (Seishin Enterprise Co. Ltd., Tokyo, Japan) at a pusher nozzle pressure and grinding nozzle pressure of 0.65 and 0.60 MPa, respectively. The ratio of peptide to excipient was 1: 400, w/w. The micronized materials were decompounded with ten-fold carrier particles (Pharmatose®) in a plastic bag for 3 min, and the obtained dry powders of peptide were stored in a vacuum desiccator until tested.
Particle size analysis:
Particle size of DPI and its dispersion were evaluated with the use of laser diffraction particle size analyzer (LMS-30, Seishin Enterprise Co. Ltd., Tokyo, Japan). DPI was subjected to dry spraying at a pressure of 0.15 MPa for effective dispersion into fine particles and carrier molecules, and then their particle size was calculated.
Scanning electron microscope:
The blends composed of fine particles and carrier was coated with gold on Quick Coater SC-701 (Sanyu Electron Co. Ltd., Tokyo, Japan). Representative scanning electron microscope images of dry powder were taken using scanning electron microscope, JEOL JSM-T220A (JEOL Ltd., Tokyo, Japan). Physicochemical evaluation of DPI using cascade impactor the dispersibility of dry powder was assessed using the cascade impactor according to the USP 23 b601N AEROSOLS. Briefly, dry powders were filled into a No. 2 hard capsule of hydroxypropyl-methylcellulose (Shionogi Qualicaps Co. Ltd., Nara, Japan), and the capsule was installed in a Jethaler® (Hitachi Unisia, Kanagawa, Japan) powder inhaler. The dry powder formulations (40 mg) were dispersed via the device with inspiration rate of 28.3 L/min for an inhalation time of 10 s×10 times, and the collection stages of the impactor (stages 0–7) were washed with 0.1% trifluoroacetic acid solution. The measurement of peptide content in each solution was conducted by RP-HPLC equipped with a fluorescence detector RF-535 (Shimadzu, Tokyo, Japan). The mobile phase of water/acetonitorile (70:30, v/v) containing 0.1% TFA was degassed and pumped through a TSK gel ODS- 120T column (particle size 5 μm, 4.6 mm i.d.×250 mm, TOSHO Tokyo, Japan) at a flow rate of 1.0 mL/min, and the column temperature was maintained at 40 °C. Animals and intratracheal administration Male Sprague–Dawley rats (9–11 weeks of age) (Japan SLC Inc., Shizuoka, Japan) were housed three per cage in the laboratory with free access to food and water, and maintained on a 12 h dark/light cycle in a room with controlled temperature (24±1 °C) and humidity (55±5%). Rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.), and then they received intratracheal administration of VIP (50, 100 μg/rat)-DPI, IK312532 (50, 100 μg/rat)-DPI or excipient-DPI. At 0.5 and 2 h after the drug administration, the rats were exsanguinated via the descending aorta under anesthesia with sodium pentobarbital, and lungs were perfused by injecting 0.9% saline into the descending aorta in the retrograde manner and removed. All procedures used in the present study were conducted according to the guidelines approved by the Institutional Animal Care and Ethical Committee of University of Shizuoka.
Tissue preparation and VIP binding assay:
The lung tissue was homogenized in 19 volumes of ice-cold 50 mM Tris–HCl buffer (pH 7.4) containing 250 mM sucrose, 5 mMMgCl2 and 0.1% bacitracin with a Polytron homogenizer, and the homogenate was centrifuged at 30,000 g for 20 min. The pellet was finally suspended in ice-cold 25 mM Tris–HCl buffer containing 5 mM MgCl2, 0.1% bacitracin and 0.2% bovine serum albumin (BSA) and used in the [125I]VIP binding assay. All steps for the tissue preparation were performed at 4 °C. The [125I]VIP binding assay was performed by a modification of the procedure described by Leroux et al. (1984). Briefly, the homogenates (20–40 μg of protein) of rat lung tissues were incubated with different concentrations (0.03–1.50 nM) of [125I] VIP in a total volume of 500 μL. Incubation was carried out for 3 h at 4 °C. The reaction was terminated by rapid filtration through What man GF/C glass fiber filters presoaked in 0.1% polyethyleneimine solution for 1 h, and filters were rinsed three times with 2 mL of ice-cold 25 mM Tris–HCl buffer containing 5 mM MgCl2 and 0.2% BSA. Tissue-bound radioactivity was determined in a gamma counter. The specific binding of [125I] VIP was determined experimentally from the difference between counts in the absence and presence of 3 μM unlabeled VIP. All assays were conducted in duplicate. Protein concentrations were measured according to the method of Lowry et al. (1951) with BSA as the standard.
Data analysis:
The analysis of binding data was performed as described previously (Yamada et al., 1980). The apparent dissociation constant (Kd) and maximal number of binding sites (Bmax) for [125I]VIP (0.03–1.50 nM) binding were estimated by Rosenthal analysis (Rosenthal, 1967) of the saturation data. The ability of VIP and IK312532 to inhibit specific [125I]VIP (0.03 nM) binding in vitro was estimated by IC50 values, which are the molar concentrations of unlabeled drugs necessary for displacing 50% of specific binding (estimated by log probit analysis). The inhibition constant, Ki was calculated from the equation, Ki=IC50 / (1+L/Kd), where L equals concentration of [125I]VIPThe data were presented as mean±S.E.M. Statistical analysis of the data was performed by one-way analysis of variance followed by Dunnett’s test for multiple comparisons. Avalue of Pb0.05 was considered significant.
Conclusion:
the application of VIP therapy inasthmatic patients has been attempted (Morice et al., 1983;Barnes and Dixon, 1984). However, the effect of VIP has beenquestioned for some reasons. It has been shown that the inhaled VIP is rapidly degraded by endogenous proteases and theintravenous administration of VIP causes cardiovascular sideeffect (Morice et al., 1983; Tam et al., 1990; Lilly et al., 1994). On the other hand, there is a report that the inhalation of syntheticVIP analogue (Ro25-1553) at doses causing a broncho dilatoryeffect does not lead to any side effect on the cardiovascularsystem (Linden et al., 2003). Thus, the long-acting VIP analoguemay be advantageous for the therapy of pulmonary diseases. Inconclusion, the present study has shown that IK312532-DPI may be a pharmacologically useful drug delivery system for theVIP therapy of pulmonary diseases such as asthma.
10. The correlation of urinary levels of albuterol and its metabolites isomers followin inhalation from a dry powder inhaler and in vitro particle size characterization:
Introduction:
Albuterol is a b2-selective adrenoceptor agonist which has been shown to exhibit considerable brocho dilatory effects. It is known that albuterol is metabolised in humans to a sulfate conjugate after oral and intravenous (I.V.) dosing [1] and the conjugate possesses little or no badrenergic activity. Albuterol and its metabolites are rapidly excreted in the urine with about 80% of the dose being recovered within 24 h [2]. About 30% of the dose is excreted in urine as unchanged albuterol 24 h after oral inhalation. Following I.V. infusion of albuterol for 2 h to volunteers, over 75% of the dose was recovered in urine within 24 h, with approximately 65% being unchanged drug and 10% metabolites. The elimination half-life of albuterol has been shown to range from 3 to 6 h after either I.V. or oral administration [1]. Albuterol has a single asymmetric carbon and is usually administered as the racemate although therapeutic activity is confined to the R-(_)-isomer with no activity observed with the S-(+)-isomer [3]. The pharmacodynamics are reported in terms of forced vital capacity (FVC), forced expiratory volume in 1 s (FEV1) and mid-expiratory flow(FEF25–75). The present study used a three-way cross-over design, with chiral analysis of the drug in urine samples to examine each enantiomer of the parent drug and its sulfate metabolite, following the inhalation of a single dose of racemic albuterol sulfate from three different formulations in 12 healthy volunteers and 12 asthmatics. The correlation between in vitro evaluation and urinary data was examined. The excretion of albuterol and its metabolites in healthy volunteers and asthmatics were explored together with factors affecting the enantioselective metabolism of the drug. The enantiomeric ratios of the parent drug and its metabolites in both healthy volunteers and asthmatics were compared. Studies were carried out to determine whether the amount of R-(_)-albuterol remaining to be excreted (ARE) correlated with the pharmacodynamic
Aerosol formulations and administration:
The formulations were prepared by mixing micronized albuterol sulfate (0.2 g) with 13.5 g of lactose carrier of different size ranges (20–60, 15–40 and 5–20 mm) in a Turbulasmixer (Basel, Switzerland) for 1 h; they were designated formulations #1, #2 and #3, respectively. Blended materials were tested for albuterol sulfate content uniformity. Then, 27.4 mg of each blend, equivalent to 400 mg drug, was weighed into each capsule. Each formulation was delivered with a device made in house [12].
Criteria for recruiting volunteers:
All volunteers gave written consent prior to participation in this study. The protocol was approved by the Faculty of Pharmaceutical Sciences Ethics Committee (Prince of Songkla University). All procedures were carried out according to the principles of good clinical practice [13]. These volunteers were all on a normal diet throughout the study.
Healthy volunteers:
Healthy volunteers (6 males and 6 females) were recruited from non-smokers, aged between 20 and 60 years, with no evidence of heart disease. Pregnant and breast-feeding females were excluded from the study. Participants were also free of kidney disease (creatinine clearance 125713 ml/min), and showed no evidence of any hepatic disease on the basis of blood chemistry tests. Lung function tests (FVC, FEV1, FEF25–75) were carried out with a Compact II Spirometer (Vitalograph, Buckingham, UK) before administration of the drug. Volunteers were excluded if their FEV1 was less than 75% of the predicted value. During the study period, volunteers were not allowed to take any other medication (including contraceptive pills).
Asthmatics:
This group of volunteers (6 males and 6 females) were recruited using the same criteria as described above for the healthy volunteers except the predicted value of FEV1 was between 50 and 75%, as specified in the American Thoracic Society criteria for diagnosis of asthma [14]. One month prior to enrolling in this study, patients did not receive either corticosteroids or bronchodilators.
The inter-conversion of albuterol enantiomers in dry powder formulations and in voided urine:
Three different formulations of dry powders were used to investigate whether any enantiomeric inter-conversion had occurred during their manufacture. These formulations were aerosolised into a twin-stage impinger at a flow rate of 60 l/min for 10 s. The drug deposited on the upper and lower stages of the twin stage impinger was analysed for the S-(+)- and R-(_)-isomers by the chiral HPLC system described above (Section 2.3). The particle size cutoff between the two stages is 6.4 mm, and this separates the particles into two populations. The stability of albuterol enantiomers was tested in voided urine which had been spiked with pure enantiomers of albuterol. Different ratios of the albuterol enantiomers (1:4, 2:3, 1:1, 3:2 and 4:1 of S-(+)/R-(_)-isomer) at a total concentration of 50 ng were spiked into 1ml urine samples and incubated at 37 1C for 24 h. This concentration was chosen so that reliable peaks for any degradation products would be produced. The enantiomeric ratios of the drug recovered from the urine were calculated and compared with the spiked concentrations in water without any extraction.
Extraction procedures and validation of the chromatographic system:
Blank urine was spiked with known amounts of racemic albuterol in the concentration range of 10–50 ng/ml. Solidphase silica cartridges were used to extract the urine samples before analysis of the drug and metabolites. The cartridges were prepared in-house by packing silica gel (40–60 mm) into Pasteur pipettes. Glass beads (2.5 mm) were placed at the tip of the Pasteur pipette to act as the bed support, followed by the silica gel (100 mg dry weight) to form a bed depth of approximately 25 mm. The cartridges were conditioned by eluting with 2ml of methanol under gravity and then washing with 2ml of water. The urine samples (1 ml) containing the drug (10–50 ng) and (–)-phenylephrine as the internal standard (100 ng) were loaded on to the cartridges which were then washed with 3ml of water before altering the polarity of the eluting solvent by washing with 2ml of methanol. Finally, the cartridges were dried for 5 min by drawing air through before eluting the drug and the internal standard using 1ml of 2%triethylamine in methanol. The eluate was evaporated to dryness with a stream of oxygen-free nitrogen at 40 1C. The residue was reconstituted with 250 ml HPLC mobile phase, and a 100 ml sample was injected onto the chromatographic column. The system was validated for accuracy, precision and linearity
Analysis of albuterol metabolites in urine samples:
A sulfatase enzyme mixture (50 ml, containing 5.3 m/ml of glucuronidase and 19.4 m/ml of arylsulfatase from Helix pomatia) (Sigma Chemicals, St. Louis, MO, USA) was added to 5ml of the urine sample followed by 1 ml of 100mM pH 5.2 acetate buffer. The mixture was incubated at 37 1C in a screw-cap container overnight. Two milliliters of 100mM pH 9.0 borate buffer was then added to neutralise the product prior to extraction and analysis as described in Section 2.5. The amount of each metabolite was calculated from the differences in the total amount of 38 T. Srichana et al. / Pulmonary Pharmacology & Therapeutics 20 (2007) 36–45 drug found in the urine before and after hydrolysis with the enzyme mixtures [16].
Data analysis:
The amount of each albuterol isomer and its metabolites in the urine for each collection period were added to give the cumulative amount excreted up to 24 h. Excretion time profiles for each albuterol isomer and its conjugated metabolites were generated both for the healthy volunteers and the asthmatics. The amount excreted during each time interval was calculated from the product of the urine volume collected and its isomer concentration. Estimates of elimination rate constants can be made by analyzing cumulative excretion data if complete urine recovery is ensured which was the case in this work. The cumulative amount excreted up to any time was obtained by adding the amount excreted in each time interval up to that time. Initially, large amounts were excreted, but as the amount of each enantiomer in the body fell, a lesser amount was excreted. The difference between the total amount excreted and amount excreted up to that time gives the amount remaining to be excreted. In practice, the value of the amount remaining to be excreted during each time was obtained by subtracting the cumulative amount excreted up to that time from the total amount of isomer excreted. A natural semilogarithmic plot of the amount remaining to be excreted versus time should give a straight line of which the slope k represents the elimination rate constant. Statistical analysis was performed using analysis of variance (ANOVA) and parameters showing significant difference between groups were further analysed using a paired t-test. Data are reported as mean7SD.
Conclusion:
In its quest for higher-performance DPIs, the pharmaceutical industry has focused very much on device technology and engineering- based approaches. By contrast, outside of academic studies, very little attention has been paid to improving the powder formulations these devices are used to deliver. However, it is important to remember that it is the performance of the system as an ensemble that is paramount for optimal therapeutic performance. It is therefore essential that the powder formulation is given the same weighting as the device in the development process. As further insight is gained into the relationship between the physicochemical and electrostatic properties of inhalation powders, and their influence on product performance, more imaginative and effective formulation strategies than have been typically applied to date can be expected in the next generation of DPI products.