1. INTRODUCTION
Plants play an important role1 in our life. Plants2 have been providing the human beings with the basic necessities of life; that is food, fuel and shelter, from the very beginning of their existence, and for their continued living.
Plants and man2 are inseparable. Because plants not only provide man the basic needs of life but also the life sustaining oxygen gas. Since man encountered diseases, got injured and wounded, faced decay of health. So, to live on earth the early man had to think about disease and its treatment at the very dawn of human intellect. And plants were the first natural substances that man could think of its use as a means of treatment of diseases. From ancient time to modern age, the human race has successfully used plants and plant products as effective therapeutic tools fighting against diseases and various other health hazards.
Again plants produce other substances by metabolic processes, such as glycosides, steroids, terpenoids, alkaloids, resins, essential oils, tannins, flavonoids, pigments etc. These are usually called secondary metabolites. Some of the compounds are harmful to animal life but some of them have proved to be tremendously effective in therapeutic doses for successful remedies for a number of diseases. Many chemical compounds of diversified nature from plants often played an important role to give a new direction for laboratory synthesis of many new classes of drug molecules.
The natural product researchers have been trying to find out the active constituents of toxic and nontoxic plants for treatment of various diseases. In modern medicine, increasing emphasis has given on pure compounds as an effective therapeutic agent and attempt has been taken to develop this by isolating, characterizing and manipulating the individual compound in plants as well as by studying certain extracts or fractions of the plant using a more holistic approach. With the advent of rapid isolation techniques, bioassay screening procedures and spectroscopic methods for structure determination the phytochemistry has become an extremely exciting and productive field in mainstream of science. Hence, it is the importance of plant and its derivatives.
Chemistry1 for utilization of medicinal plants started to develop their activity as a therapeutic agent. In the recent decades natural pesticides, antibiotics vitamins and hormones are the follow up results of the researchers of chemistry of natural products. Scientists are now working together to find out new drugs from plants for incurable diseases likes diabetes, cancer and AIDS. So, it will not be at all surprising that the chemistry of natural products will lead to new contributions to therapy of the controlled diseases.
Ordinary people do not know the harmful and useful effects of the compounds stored in the plants; they also do not know what the side effects of such plant materials are. So the chemists have a great responsibility to find out the active ingredients, which are effective against various diseases and also to identify the toxic ingredients (if any).
1.2 MEDICINAL IMPORTANCE OF PLANT MATERIALS
A medicinal plants is any plant which, in one or more of its organs, contains substances that can be used for therapeutic purposes or which are precursors for synthesis of useful drugs. The consultive group on medicinal plants of World Health Organization (WHO) has given this definition. Unfortunately this definition of the WHO group includes only the medicinal plants whose therapeutic properties and chemical constituents have been established scientifically. But it does not take into consideration the vast majority of the medicinal plants, which have not yet been subjected to through scientific studies. These medicinal plants have been used in traditional medicine for hundreds of years with reputation as efficacious remedies although there may not be scientific data to substantiate their efficiency.
Thus plants that posses therapeutic properties or exert beneficial pharmacological effects on the animal body are designated as medicinal plants. Although there are no apparent morphological characteristics in the medicinal plants that make them distinct from other plants growing with them, yet they possess some special qualities or virtues that make them medicinally important. It has now been established that the plants which naturally synthesize and accumulate some secondary metabolites, like alkaloids, sterols, terpenes, flavonoids, saponins oils, etc and contain minerals and vitamins, possess medicinal properties.
Today there are4 at least 120 distinct chemical substances from plants that are considered as important drugs currently in use in one or more countries in the world. Several of the drugs sold today are simple synthetic modifications or copies of the naturally obtained substances. The original plants substance or chemical name is shown under the “ Drug ” column rather than the finished patented drug name. For example, many years ago a plant chemical was discovered in a tropical plant, Cephaelis ipecacuanha, and the chemical was named emetine. A drug was developed from this plant chemical called Ipecac, which was used for many years to induce vomiting mostly if someone accidentally swallowed a poisonous or harmful substance. Ipecac can still be found in pharmacies in many third world countries but has been mostly replaced by other drugs in the United States. Another example of this is the plant chemical named Taxol shown in the drug column below. Taxol is the name of a compound discovered from a plant. A pharmaceutical company copied this chemical and patented a drug named PaclitaxelTM which is used in various types of tumers today in the U.S. and many other countries.
The chemical substances derived from plants are sold as drugs worldwide but not in all countries. Some European countries regulate herbal substances and products differently than in the United States. Many European countries, including Germany, regulate herbal products as drugs and pharmaceutical companies prepare plant-based drugs simply by extracting out the active chemicals from the plants. A good example is the plant substance/drug shown below, cynarin. Cynarin is a plant chemical found in the common artichoke (Cynara scolymus). In Germany, a cynarin drug is sold for liver problems and hypertension which is simply this one chemical extracted from the artichoke plant or a plant extract which has been standardized to contain a specific milligram amount of this one chemical. Pharmaceutical companies, sold in pharmacies in Germany, manufacture these products and a doctor’s prescription is required to purchase them. In the United States artichoke extracts are available as natural products and sold in health food stores. Some products are even standardized to contain a specific amount of the cynarin chemical. We can purchase these natural and standardized extracts over the counter without a prescription and we could not go to a pharmacy in the U.S. and obtain a cynarin drug with a prescription. Another similar example is the plant chemical found in the milk thistle plant and natural milk thistle extracts standardized to contain specific amounts of silymarin are found in just about every health food store in the United States. However in Germany, silymarin drugs and milk thistle standardized extracts are sold only in pharmacies and require a doctor’s prescription for liver problems.
Some of the drug/chemicals shown below are still sold as plant based drugs requiring the processing of the actual plant material. Others have been chemically copied or synthesized by laboratories and no plant materials are used in the manufacture of the drug. A good example of this is the plant chemical quinine, which was discovered in a rainforest tree (Cinchona ledgeriana) over 100 years ago. For many years the quinine chemical was extracted from the bark of this tee and processed into pills to treat malaria. Then a scientist was able to synthesize or copy this plant alkaloid into a chemical drug without using the original tree bark for manufacturing the drug. Today, all quinine drugs sold are manufactured chemically without the use of any tree bark. However, another chemical in the tree called quinine that was found to be useful for various heart conditions couldn’t be completely copied in the laboratory and the tree bark is still harvested and used to extract this plant chemical from it. Quinidine extracted from the bark is still used today to produce quinidine-based drugs sold in pharmacies containing bark-extracted quinidine: CardioquinTM, Quinaglute Dura-tabsTM, Quinidex ExtentabsTM and Quin-ReleaseTM.
Table-1.1: IMPORTANT DRUGS/CHEMICALS FROM PLANT SOURCE AND THEIR ACTIONS/CHEMICAL USES 4
Drug/Chemical | Action/Clinical use | Plant source |
Betulinic acid | Anticanceous | Betula alba |
Camptothecin | Anticancerous | Camptotheca acuminata |
Chymopapain | Proteolytic, mucolytic | Carica papaya |
Cissampeline | Skeletal muscle relaxant | Cissampelos pareira |
Colchiceine amide | Antitumer agent | Colchicum autumnale |
Colchicine | Antitumer agent, antigout | Colchicum autumnale |
Curcumin | Choleretic | Curcuma longa |
Cynarin | Choleretic | Cynara scolymus |
Danthron | Laxative | Cassia species |
L-Dopa | Anti-parkinsonism | Mucuna sp. |
Etoposide | Antitumer agent | Podophyllum peltatum |
Glaucarubin | Amoebicide | Simarouba glauca |
Glycyrrhizin | Sweetener, Addison’s diseases | Glycyrrhiza glabra |
Hesperidin | Capillary fragility | Citrus species |
Irinotecan | Anticancer, antitumor agent | Camptotheca acuminata |
Lapachol | Anticancer, antitumor | Tabebuia sp. |
Menthol | Rubefacient | Mentha species |
Papain | Proteolytic, mucolytic | Carcica papaya |
Pilocarpine | Parasympathomimetic | Pilocarpus jaborandi |
Podophyllotoxin | Antitumor anticancer agent | Podophyllum peltatum |
Quinidine | Antiarrhythmic | Cinchona ledgeriana |
Quinine | Antimalarial, antipyretic | Cinchona ledgeriana |
Rutin | Capillary fragility | Citrus species |
Sennosides A, B | Laxative | Cassia species |
Stevioside | Sweetner | Stevia rebaudiana |
Taxol | Antitumor agent | Taxus bevifolia |
Teniposide | Antitumor agent | Podophyllum peltanum |
α-Tetrahydroxcannabinol (THC) | Antiemetic, decrease ocular tension | Cannabis sativa |
Theobromine | Diuretic, vasodilator | Theobroma cacao |
Theophylline | Diuetic, brochodilator | Theobroma cacao and others |
Topotecan | Antitumor, anticancer agent | Camptotheca acuminata |
Trichosanthin | Abortifacient | Trichosanthes kirilowii |
Tubocurarine | Skeletal muscle relaxant | Chondodendron tomentosum |
Vasicine | Cerebral stimulant | Vinca minir |
Vinblastine | Antitumor, antileukemic agent | Catharanthus roseus |
Vincristine | Antitumor, Antileukemic agent | Catharanthus roseus |
STRUCTURE OF SOME OF THE MODERN DRUGS ORIGINATED FROM NATURAL PRODUCTS ARE GIVEN BELOW
1.3 THE FAMILY POLYGONACEAE
The family Polygonaceae consists of 40 genera comprising about 750 species. The members of this family are annual or perennial herbs; rarely shrubs and very rarely trees5. They are widely distributed throughout the world and divided into two ‘tribes’ called Polygoneae and Persicarieae6. The term ‘tribes’ represent a category usually comprising of two or more genera.
1.4 THE GENUS POLYGONUM L.
The members of the genus Polygonum are annual or perennial herbs, sometimes woody and belong to the tribe Polygoneae under the family Polygonaceae. It is the largest genus of the family with about 300 species of cosmopolitan distribution7. It is especially abundant in temperate regions. In Bangladesh, the genus Polygonum L. is represented by 20 species and 9 varieties under 5 sections (Table A-2)8. Most of them are riparian and forms characteristic marsh vegetation in the lowland areas. They grow in very huge quantities, and some are commonly used by the rural people as pesticides, as potherbs and for a variety of ailments that affect men and animals8.
1.5 BOTANICAL CHARACTERISTICS5 OF THE GENUS POLYGONUM L.
Herbs (rarely shrubs), usually erect, rarely climbing.
Leaves: The leaves are simple, alternate, mostly entire, rarely lobed, base, attached to stipular sheath called ochrea.
Flowers: The flowers are hermaphrodite, small or minute, axillary or terminal, bisexual, pedicel joined in the middle or under the perianth.
Perianth: Green or coloured, 4-5- (rarely 3-) cleth, the 2 outer segments usually smaller than the others.
Stamens: 5-8 (rarely 1-4), perigynous; filaments filaments filiform, usually dilated below or alternate with the lobes of an annular or glandular disk; anothers 2-called; cells discreate; connective small.
Tepals: 4-5 (rarely 3 or 6), membranous or fleshy in fruits.
Antehrs: 2-called, the cells distinct, joined by a small connective.
Ovary: 2-faced or 3-gonous, avules solitary, usually stipitate, styles 2 or 3, free or slightly connate below, stigma capitate.
Fruits: A compressed or 3-gonous nutlet with obtuse of acute angles, coyered by the persistent perianth, pericap usually hard, shining or dull.
Nuts: Lenticular of trigonous, sometimes gobular, rarely tetra, penta or hexagonous, smooth or recticulate.
Seeds: Albumions, embrys lateral or eccentric, radicle long, cotyledons feat or plaited, accumbent or incumbet.
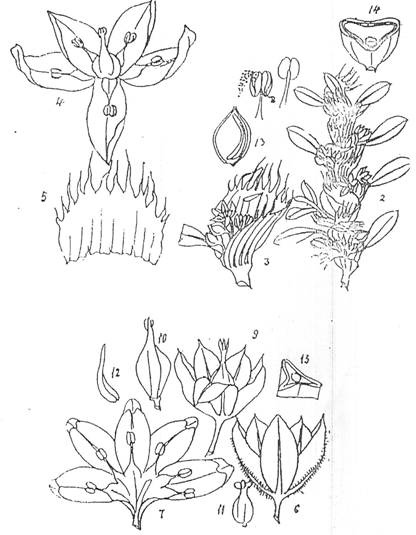
1.6 TAXONOMIC CHARACTERISTIC5 OF THE SPECIES POLYGONUM
A diffusely branched, prostrate herb, often with a woody rootstock, branches terete, striate.
Leaves: Leaves are almost sessile, narrow, up to 3.0 X0.2 cm, stipules hyaline, lacerate to the middle, fimbriate.
Flowers: Flowers are pink, axillary, 3-4 per axil, pedicellafe, not exerted.
Perianth: 2.5 mm long or less, divided nearly to the base.
Stamens: 5, c. 0.3-0.4 mm, filament with feat and dilated base, white.
Tepals: 5, single veined, greenish- with or light pink inside, at least the outer two acute, and the outermost longer, corrugate dorsally.
Athers: White or light pink.
Ovary: Trigonous, c, 0.3 mm, styles 3, very short, stigmas 3.
Nuts: 3-gonous, c.1.0 0.5 mm, smooth, black, shining.
1.7 SPECIES OF POLYGONUM GENUS AVAILABLE IN BANGLADESH
The members of the family Polygonaceae are chiefly found in the north temperate zone of the world. Only a few genera are tropical e.g. Coccoliba is confined to tropical and sub-tropical America. Oxyria digyna and Koenigia islandica are found in the colder parts of Northern hemisphere. A few genera are widely distributed, e.g. Polygonum is worldwide in its distribution and found in all the five continents. In our country the family is represented by several important genera such as, Polygonum, Antigonum, Rumex, Fagopyrum, Muehlenbeekia, Coccoloba etc. The species of Polygonum geneus abundantly grow in Bangladesh and most of them are found to grow throughtly the country. Table–1.2 represents a list of some species of Polygonum genus available in Bangladesh and their location in other countries.
Table-1.2: LIST OF SOME POLYGONUM SPECIES AVAILABLE IN BANGLADESH AND THEIR LOCATIONS IN OTHER COUNTRIES
| Name of species | Locations |
| Polygonum effusum | Afganistan, Pakistan, India |
| Polygonum chinense L. | India, Nepal, China, Japan |
| Polygonum orentale L. | Bangladesh, India, Nepal, Mayanmer, Thailand, Turkistan,Chaina, Japan, Indonesia |
| Polygonum sagittatum L. | India, Nepal, Chaina, Mayanmer, Australia, Europe, Africa, America |
| Polygonum hydropiper L. | Bangladesh, India, Indonesia, Europe, North Africa, Australia, North America |
| Polygonum strigosum R. Br. | Bangladesh, India |
| Polygonum lapathifolium L. | Bangladesh, India |
| Polygonum flaccidum | Bangladesh, India |
| Polygonum plebejum R. Br. | Bangladesh, India, Pakistan, Nepal, Bhutan, Burma, Sri Lanka, China, Taiwan, Philippines, Indonesia, Malaysia, Australia, Africa |
| Polygonum limbatum Meissn. | Bangladesh, India |
| Polygonum perfoliatum L. | Bangladesh, India |
| Polygonum assamicum Meissn. | Bangladesh, India |
| Polygonum praetermissum Hook. | Bangladesh, India |
| Polygonum barbatum L. | Bangladesh |
| Polygonum glabrum Willd. | Bangladesh |
| Polygonum posumba Buch-Ham. | Bangladesh, India |
| Polygonum tomentosum Willd. | Bangladesh, India |
| Polygonum viscosum Ham. | Bangladesh, India |
| Polygonum fagopyrum L. | Bangladesh |
| Polygonum minus Huds. | Bangladesh, India |
| Polygonum macranthum Meissn | Bangladesh, India |
| Polygonum microcephalum D. Don. | Bangladesh, India |
1.8 ECONOMICAL IMPORTANCE OF POLYGONACE
Some plants of this family are ornamental, some of this is used as food and some possess medicinal properties. A list of few important plants is given below:
Antigonon leptopus; Eng.- Coral creeper. This is a large climbing shrub with beautiful pink flowers. It is native of South America. Grown as an ornamental in the gardens.
Muehlenbeckia platycladus; Syn. Coccoloba platyclada; Eng.- Centipede plant. This is a small shrub. It is native of the Solomon Islands. It is grown as an ornamental. It possesses flat, green phylloclades. The fruit is astringent.
Fagopyrum tataricum; Eng.- Tatary buckwheat, India wheat; Verna.- Kaspat, Paphra- it is grown throughout the Himalayas. The grains are usedas food.
Fagopyrum cymosum; Eng.- Perennial buckwheat; Verna.- Banogol- This is a tall herb, found in the temperate Himalayas and the Khasia hills. The leaves are used as vegetable.
Polygonum plebejum; Verna.- Rainphul- The roots possess medicinal properties and are used in the treatment of pneumonia.
Polygonum bristorta; Eng.- Snakeweed. It is a perennial herb. The roots are used for tanning.
Polygonum chinense; Verna.- Ameta- This is an undershurb, grown as an ornamental.
Polygonum fagopyrum; Syn. Fagopyrum esculentum; Eng.- Buckwheat; Verna.- Kutu- This is a native of Central of Central Asia, but now extensively cultivated in the hills of Northern India. The grains (seeds) are edible and used as food.
Rheum emodi; Eng.- Himalayan rhubarb; Verna- Revand chini- this is a herb, found in the sub-alpine. Himalayas. The roots and rhizomes are the source of a drug, which is used as alaxative, tonic and purgative.
Rheum rhabarbarum; Syn. Rheum undulatum; Eng.- Bucharian rhubarb. The leaves are eaten as vegetable.
Rheum rhaponticum; Eng.- Garden rhubarb. This is a large shrub, cultivated in Tamil Nadu and Bengal. The thick leafstalks are eaten as vegetable.
Rumex acetosa; Eng.-Garden sorrel; Verna- Chuka- This is a herb, commonly found in the Western Himalayas from Kashmir to Kumaon. The leaves are consumed as vegetable.
Rumex crispus; Eng.- Curled dock; Verna.- Chukkah- The leaves are eaten as vegetable.
Rumex dentatus; Verna- Lalbibi, Khatpalak- this is found in North western Indid, Kumaon, South India and the Western Ghats.
The roots yield a red dye.Rumex hastatus; Verna.- Bhilmora- The leaves are used as condiment.
Rumex hymeirosepalus; Eng.- Canaigre. This has been introduced from the United States. The tuberous roots are used as atan.
Rumex vasicurium; Eng.- Bladderdock; Verna.- Chuka- Cultivated all over India. The leaves are eaten as vegetable. The leaves are also used in snakebite.
1.9 THE PLANT POLYGONUM PLEBEJUM
Polygonum plebejum is a much-branched annual herb and grows almost all over Bangladesh. It is commonly fund in dried up ponds, ditches, riverbanks, cultivated fields and low lands. Locally it is called ‘Khudi-Bishkatali’ or ‘Mechhua Sank’. It is also distributed throughout tropical India and sometimes ascending the Himalayan up to 7000 ft, Pakistan, Bhutan, Burma, Sri Lanka, China, Taiwan, Philippines, Indonesia, Malaysia, Australia and Africa9.
1.10 THE CLASSIFICATION OF POLYGONUM PLEBEGUM 10
Kindom: Plantae (Standard group)
Tracheobionta (Sub group)
Phylum: Magnoliophyta
Class: Magnoliopsida
Caryophyllidae (Sub group)
Order: Polygonales
Family: Polygonaceae
Genus: Polygonum
Species: Polygonum plebejum

Fig- 1.2: Polygonum plebejum
1.11 MEDICINAL IMPORTANCE OF POLYGONUM GENUS
A number of species of the genus Polygonum are being used5 ethno medically in many countries for various kinds of diseases. Some of them are described:
The hurbs and leaves of Polygonum hydropiper are stimulant, diuretic, emmenagougue and are used principally in Europe for obstructions of the menses and amenorrhoea. The root is stimulating, bitter and tonic. The juice of this plant is used for the treatment of icing and is used as a diuretic, carminative and anthelmintic.
The seeds of Polygonum aviculare is used as laxative, diuretic and used for remedy of pain in the stomach and bladder. In Kashmir, the seeds are used as an emetic and purgative5. The herb is considered astringent in Europe and its infusion has been found beneficial in diarrhoea and children’s summer complaints. It is used in Malaya for gonorrhoea.
The roots of Polygonm barbatum is used as an astringent and cooling remedy. A decoction of the leaves and stalks is said tobe used as a curative wash for ulcers.
The plant Polygonum persicaria is considered astringnt and used as avulnerary and lithontriptic. The juice of this plant is used to relieve the pain of Muscles.
Polygonum orientale and Polygonum chineese have good tonic and vulnerary properties, the latter species has ant scorbutic properties too.
The roots of Polygonum viviparum is a useful astringent and its decoction makes an excellent gargle for curing score-throat and spongy, gums. It is an excellent lotion for ulcers. According to Yunani, the root is tonic to chest and lungs. It is also used for treatment of piles, old diarrhoea, rhintis, vomiting, biliousness, chronic bronchitis, wounds and gripping in the abdomen.
The dried powder of the plant Polygonum plebejum is used internally in pneumonia. In India, the Santals use the root of this plant to get rid of bowel complaints.
1.12 BIOLOGICAL ACTIVITIES OF POLYGONUM PLEBEJUM
Human diseases of bacterial origin are varied and are of different types. In the humid tropic land of Bangladesh, Cholera, typhoid, dysentery, septicaemia and diptheria are only a few of the killer diseases reigning during the last quarter of the 20th century. Plants and plant material have been tried to relieve human ailments even from the prehistoric era. Still there is a desperate need for the study on the antibacterial activities of plants to control the common diseases.
Although a large number of species of Polygonum genus are being used ethnomedicaly for different kinds of diseases throughout the world, almost no chemical or biological investigation has been done on Polygonum plebejum, but recently antibacterial activity of five species (P. flaccidum, P. lapathifolium, P. orientale, P. plebegum, P. strigosum) of family Polygonacea has been carried out. This investigation12 reported that Polygonum plebejum has significant antibacterial activity against both Gram positive and Gram-negative bacteria but alcoholic extract are inactive to these bacteria. It has also reported that 10% oily extract of this plant was also active against Salmonella paratyphi A. and Shigella flexneri.
1.13 CHEMICAL INVESTIGATION10 ON POLYGONUM GENUS
The literature survey showed that limited phytochemical work has been done on Polygonum plebejum. However, report of investigation on a few other species of this genus has been found. These are cited bellow:
The isolation of four diesters of 2,3-dihydroxy isodimenitol (1-4) from Polygonum glabrumin has been reported.
A stibene glycoside identified as 2,3,4,5-tetrehydroxy stilbene-O-β-D glucoside (5) has been isolated and other two-stilbene glycoside gallates (6,7) identified as monogaloye esters of 2,3,5,4/-tetrahydrostilbene-2-O-D-glucopyranoside (7) have been isolated and characterized along with proanthocyanidins from Polygonum multiflorum.
Four known flavonoids from Polygonum sieblolide and Polygonum filiforwe have been reported. The latter species yielded the flavonoid glycoside identified as quercetin-3-rhamnoside-2”-gallate (8).
The hydrolysate of foliage of Polygonum aviculare was analysed and found to contain 9.54% amino acids. The presence of 16 amino acids of which six amino acids are essential, e.g. methionine, proline, serine, threonene, tyrosine and phenyl alamine has been reported.
Isolation of quercetin –3-methylether (9), quercetin-3-glycoside (10), quercetin-3-rutinoside (11), kaempferol-3-glucoside (12), kaempferol-3-methyl ether (13), kaempferol-3-glucoside and chrysine along with β-sitosterol-p-hydroxy benzoic acid, vanillic acid, gallic acid (14) and protocatechuic acid (15) has been reported from Polygonum cognatum.
Investigation on the leaves of 28 species belonging to the family Polygonaceae has been carried out and the isolation of 33 flavonoids has been reported.
The types of flavonoids were quercetin glycosides, 3-O-rhamnoside, 3-O-glucurinide, methylated flavonols and c-glycoflavones.
A report on isolation of flavonoid glycosides from aerial parts of Polygonum lapothifolium has been published. Six of these flavonoids were identified as quercetin-3-O-β-D glucopyranoside (10), quercetin-3-O-β-D-galactopyranoside (16), quercetin-3-O-β-D-glucoside-2”-gallate (17), quercetin-3-O-α-L-arabofuranoside,Kaempferol-3-O-β-Dgalactopyranoside (18) and Kaempferol-3-O-β-D- glucoside-2”-gallate (19). Quercetin and its glycosides with arabinose and xylose have been characterized from Polygonum amphibium.
A report on isolation13 of polygodial (20) from Polygonum plebejum has been published. It was also isolated from Polygonum hydropiper.
Structures of the isolated and characterized compounds mentioned above are listed below:
SOME OTHER IMPORTANT COMPOUND OF THE GRNUS ARE REPORTED HERE 17
Name Structure
Anibadimer A
Astragalin;
2”-O-(3,4,5-Trihydroxybenzoyl)
Avicularin
1,3-Bis (4-methoxy-2-oxo-6-pyranyl)-2,4-diphenylcyclobutane; (1α, 2α, 3β, 4β)
3,6-Digalloylglucose; D-pyranose
3,4-Dihydro-5,7-dihydroxy-4-(4-hydroxyphenyl)-2H-1-benzopyran-2-one; (S)
3,4-Dihydro-5, 7-dihydroxy-4- (4-hydroxyphenyl)-2H-1-benzopyran-2-one; (S), 7-Me ether
3,4-Dihydro-5,7-dihydroxy-4-94-hydroxyphenyl)-2H-1-benzopyran-2-one; (S), 4’-Me ether
1,3-Dihydroxy-6,7-dimethylxanthone; 1-O-β-D-Glucosopyranoside
1-(3,5-Dihydroxyphenyl)-2-(4-hydroxyphenyl) ethelene; (E)
1-(3,5-Dihydroxyphenyl)-2-(4-hydroxyphenyl) ethylene; (E), 3-O-β-D-Glucopyranoside
Llagic acid, 1NN; 3,8-Di-Me ether,
2-O-α-L-rhamnopyranoside
Ellagic acid, INN; 3,8-Di-Me ether, 2-O-[β-D-glucopyranuronosyl-(1-4)-α-L-arabinopyranosyl-1(1-6)-β-D-glucopyranoside]
6-Galloyglucose
3,4,5,5,6,7-Hexahydroxyflavone; 6,7-Methylene, 3,4,5,5-tetra-Me ether
3-Hydroxy-5-phenylpentanoic acid; (R), O-β-D-Glucopyranoside
3,4,4,5,9,9-Hexahydroxy-2, 7-cyclolignan; (7s,8R,8R),3,5-Di-Me ether, 9-O-α-L-rhamnopyranoside
1-(4-hydroxyphenyl0-2-(2,3,5-trihydroxyphenyl) ethylene; 2-O-β-D-Glucopyranoside
Isoquercitin; 2-O-(3,4,5-Trihydroxybenzoyl)
Lapathinol
3,3,4,5,5,6,7,8-Octahydroxyflavone; 3,45,6,7,8-hexa-Me ether
3,3,4,5,5,6,7,8-Octahydroxyflavone; 3,4,5,8-Tetra-Me,6,7-methylene ether
Orientin
3,3,4,5,7-Pentahydroxyflavan; (2S, 3S)
1.14 STEROIDS
Compounds which produce Diel’s hydrocarbon on selinium dehydrogenition (3′-methyl-1, 2 cyclopenteno-phenanthrne) are known as steroids14.
Diel’s hydrocarbon
Steroids contain four rings fused together. Among the four rings three are six membered and other one is five membered.
The precursor of steroids in biological system is squalene, a triterpene that was originally produced by joining of six isopentenyl pyrophosphate.
Many hormones, bile acids and vitamins have this steroidal structure. Usual birth control pill from women contains steroidal hormones. Steroids are extractible by lipophilic solvents like chloroform, petroleum ether etc.
1.15 BIOSYNTHESIS OF STEROIDS
Steroids15, 16 are not triterpenes as they possess C27-C29 skeletons; rather than a C30 skeleton, although both of them are derived from the same C30 precursor squalene. This squalene is derived from tow farnesyl-pyrophosphate units, upon joining each other in the unusual head to head’ fashion.
Squalene
Cyclisation of squalene-2,3-epoxide in a chair-boat-chair-boat conformation and by a subsequent sequence of rearrangements leads to lanosterol, cycloartenol etc. Desmosterol is formed from lanosterol by a sequence of modification reactions. -sitosterol and stigmasterol are fomed by the addition of extra carbon atoms to the side chain of desmosterol in plants.
Scheme2: Biosynthesis of phytosterols
2.1 METHODOLOGY
2.1.1 GENERAL METHODS
2.1.1.1 DISTILLATION OF SOLVENTS
Analytical or laboratory grade solvents and chemicals used in the experiments. All solvents and reagent used in the experiments were produced from E. Merk (Germany), BDH (England). The commercial grade solvents (ethyl acetate, chloroform, methanol, dichloromethane) were distilled before use. Petroleum ether (b.p. 40o-60oC & 60o-80oC) was obtained by distilling petrol. All the solvents were distilled in a glass distillation set before uses. Distilled solvents were throughout the investigation.
2.1.1.2. EVAPORATION
All experiments were carried out under reduced pressure with the help of rotary vaccum evaporator at bath temperatures not exceeding 40o.
2.1.1.3 PREPARATION OF REAGENTS
2.1.1.3.1 VANILLIN-SULFURIC ACID REAGENT
Sulfuric acid (400 mL) and absolute alcohol (100 mL) were mixed in a beaker (kept in ice bath), Vanillin (0.25 g) was added to the mixture of alcohol and sulfuric acid, cooled and used for spraying the TLC plates.
2.1.1.3.2 DRAGENDORFF’S REAGENT
Solution A: Bismath subnitrate (0.6 g) was dissolved in water (2 mL)
Solution B: Potassium iodide (o.6 g) was dissolved in water (10 mL)
These two solutions were mixed and 22 mL of dilute HCl (7 mL of conc HCl + 15 mL water) was added to it. Then the mixture was diluted with water (400 mL).
2.1.1.4 CHROMATOGRAPHIC TECHNIQUES
Two types of chromatographic techniques ere used such as thin layer chromatography (TLC) and vaccum liquid chromatography (VLC).
2.1.1.4.1 THIN LAYER CHROMATOGRAPHY (TLC)17
Two types of TLC plates were used through out the experiment:
Precoated TLC plates: 0.2 mm thin coatings of silica gel on glass or aluminium sheets were used.
Manually prepared silica gel coated glass plates were used.
2.1.1.4.2 PREPARATION OF PLATES
Thin layer chromatographic plates were prepared by spreading a film of an aqueous slurry (gel: water = 1:2 w/v ) of silica gel G-60 PF254 ( E. Merck 7731) over the entire surface of the glass plates ( 6cm x 12cm ) by means of spreader. This thickness of the silica gel layer was 0.2 mm. The plates were dried the air and finally activated by heating at 110 oC for 1 hour followed by cooling at room temperature for few hours.
2.1.1.4.3 SAMPLE APPLICATION (SPOTTING THE PLATES)
The TLC plates were spotted with a small amount of the crude extract by using a narrow capillary. The capillary was either acetone or ethanol before each sample was applied.
2.1.1.4.4 SOLVENT SYSTEMS
The solvents of different polarity used for TLC are given below:
Petroleum ether
Dichloromethane
Ethyl acetate
Methanol
Petroleum ether: Dichloromethane (in different ratio)
Petroleum ether: Ethyl acetate (in different ratio)
Dichloromethane: Dichloromethane (in different ratio)
Dichloromethane: Methanol (in different ratio)
2.1.1.4.5 PREPARATION OF TLC TANK
The ascending technique in glass jars or tanks were used to develop TLC plates. A suitable solvent system was poured into glass jar or tank in a sufficient amount. The tank was then covered with a lid and kept for a certain period allowing it to achieve saturation. A filter paper was usually introduced into the tank to promote the saturation process. The solvent at the bottom of the tank must not be above the line of the spot where the sample solution was applied to the plate. As the solvent rises upward, the plate becomes moistened. The plate was then taken out and dried. The solvent front was not allowed to travel beyond the end of the silica-coated surface.
2.1.1.4.6 DETECTION OF SPOTS
For the location of the separated components, the plates were examined by the following methods:
Examination under UV lights in different wavelength, 254 and 350 nm.
The plates were exposed to iodine vapor for several minutes.
The plates were sprayed with vanillin-sulfuric acid reagent (1.0%) followed by heating in an oven at 120 oC for 10-15 minutes.
2.1.1.4.7 The Rf VALUE
Retardation factor (Rf) is the ratio of the distance the compound travel to the distance the solvent front moves.
Usually, the Rf value is constant for any given compound and it corresponds to a physical property of that compound18.
2.1.1.5 VACUM LIQUID CHROMATOGRAPHY
The concept of vacuum liquid chromatography is a relatively recent development in the field of chromatography separation. It is a column chromatography under reduced pressure and the column is packed with TLC grade silica gel. This technique was used for fractionating the crude extracts in order either pure compound or a mixture with a small number of compounds. A glass column of about 40 cm length having a system fitted with a water pump and collecting tube at the bottom was taken (fig- ). Silica gel (60 GF254) was used as adsorbent. This slurry was packed into hard cake under an applied vacuum to give a column 3.15 cm in diameter and about 7.0 cm in height. The sample mixture or extract which to be separated, was dissolved in a minimum volume of suitable solvent. Then small amount of silica gel (around twice of the amount of sample) was mixed with sample solution. After through mixing, the solvent of the mixture of sample in silica gel was evaporated using rotary evaporator. To ensure complete removal of solvent, the sample-silica gel mixture was kept in a vacuum dessicator. After drying, the mixture was converted into the powder by mortar. The powder was then poured on the top of the packed column to from a layer. Gradient elution with increasing polarity of eluting solvent was used until the more polar component was eluted. These elutes were collected manually in the fraction of the different amounts and ten the individual fractions were monitored by TLC.
Fig-2.2: Vacuum Liquid Chromatography (VLC) apparatus
2.1.1.6 CRYSTALLIZATION
Crystallization was employed as a final purification process. A solution of the compound in a minimum volume of the solvent in which it is soluble was prepared in hot condition. It was then left for crystallization. Some time, a mixture of solvents was also used.
2.1.1.7 SPECTROSCOPIC TECHNIQUES
2.1.1.7.1 ULTRA-VIOLET SPECTROSCOPY (UV)
Ultra-violet spectra were recorded using the Shimadzu UV-160A spectrophotometer. A small amount of the samples of different compounds were dissolved in suitable solvent separately. The solution was taken in quartz cell (1cm x 1 cm) to record the spectrum. The main brands (λ max) were recorded as wavelength (nm).
2.1.1.7.2 INFRA-RED SPECTROSCOPY (IR)
A Shimadzu IR-470 spectrophotometer was used for recording Infra-red spectrum. Major bands (ν max) were recorded in wave number (cm-1) as KBr pellets.
2.1.1.7.3 NUCLEAR MAGNETIC RESONANCE (NMR) SPECTROSCOPY
1H-NMR (400 MHz) and 13C-NMR (100MHz) spectra of pure sample were recorded by using high resolution NMR (400 MHz) spectrometer. The spectra were record using CDCl3 with tetra methyl silane (TMS) as standard reference.
2.1.1.8 MELTING POINT APARATUS
Melting point (m.p) was determined by using an electro thermal melting point apparatus (Mel-temp, OGAWA SEIKICO, and Japan).
2.2 EXPERIMENTAL
2.2.1 INVESTIGATION ON POLYGONUM PLEBEJUM
2.2.1.1 COLLECTION OF PLANTS
The plant Polygonum plebejum was collected from Mohammadpur area and was identified from the department of Botany, Dhaka University. The collected fresh plants were cleaned thoroughly. The plant materials (leaves and stems) were separated from its roots and dried under mild sunlight and then at 40 oC in an oven. Afterwards the plants were powdered in a grinding machine (~200 meshes). The powder was used throughout the investigation.
2.2.1.2 PHYTOCHEMICAL SCREENING OF THE PLANTS
Chemical tests were carried out on the aqueous extract and on the powdered specimens using standard procedures to identify the constituents 19,20,21.
2.2.1.2.1 QUALITATIVE DETERMINATION
TEST FOR TANNINS
The dried powder samples (~0.5 g) was boiled in water (20 ml) in a test tube and then filtered. A few drops of ferric chloride solution (0.1%w/v) were added and brownish green or a blue-black coloration was observed.
TEST FOR PHLOBATANNINS
Deposition of a red precipitate (on boiling) extract of each plant sample was boiled with aqueous hydrochloric acid (1% w/v) indicated the presence of phlobatannins.
TEST FOR SAPONIN
The powdered sample (~2g) was boiled in distilled water (20 ml) in a water bath and filtered. The filtrate (10 ml) was mixed with distilled water (5 ml) and shaken vigorously for a stable persistent froth. The frothing was mixed with olive oil (3 drops) and shaken vigorously, and then the formation of emulsion was observed.
TEST FOR FLAVONOIDS
Three methods were used to determine the presence of flavonoids in the plant sample-
a) Dilute ammonia solution (5 ml) was added to a portion of the aqueous filtrate of each plant extract followed by addition of concentrated H2SO4.
b) Few drops of aluminium ion (Al3+) solution (1%) were added to a portion of each filtrate. A yellow colouration each observed indicating the presence of flavonoids.
c) A portion of the powdered plant sample each case was heated with ethyl acetate (10 ml) over a steam bath for 3 min. The mixture was filtered and the filtrate (4ml) was shaken with dilute ammonia solution (1 ml). A yellow colouration was observed indicating a positive test for flavonoids.
TEST FOR STEROIDS
Acetic anhydride (2 ml) was added to ethanolic extract (0.5 g) of each sample with H2SO4 acid (2 ml). The colour changed from violet to blue or green in some samples indicating the presence of steroids.
TEST FOR TERPENOIDS (SALKOWSKI TEST)
Each extract (5 ml) was mixed in chloroform (2 ml), and concentrated H2SO4 acid (3 ml) was carefully added to form a layer. A reddish brown colouration at the interface was formed to show positive test for the presence of terpenoids.
TEST FOR CARDIAC GLYCOSIDES (KELLER-KILLANI TEST)
Each extracted (5 ml) was treated with glacial acetic acid (2 ml) containing ferric chloride solution (1 drop). This was underlayed with concentrated H2SO4 acid (1 ml). A brown ring at the interface indicates a deoxysugar characteristic of cardinolides. A violet ring may appear bellow the brown ring, while in the acetic acid layer, greenish ring may from just gradually throughout thin layer.
Table-2.1: Qualitative analysis of the phytochemical of the medicinal plants
Plants Alkaloid Tannin Saponin Flavonoid Steroid Terpenoid Cardiac glycoside
Polygonum plebejum + + + + + + +
2.2.1.2.2 QUANTITATIVE DETERMINATION OF THE CHEMICAL CONSTITUENTS
ALKALOID DETERMINATION 20
Each sample (0.5 g) was taken into a conical flask (100 ml) and acetic acid (20 ml in 10%) ethanol was added. It was covered and allowed to stand for 12 hours. This was filtered and the extract was concentrated on water bath to one-quarter of the original volume. Concentrated ammonium hydroxide was added drop wise to the extract until the precipitation was complete. The whole solution was allowed to stand and the precipitate was collected and washed with dilute ammonium hydroxide solution and then filtered. The residue is the alkaloid, which was dried and weighed. The results are given in Table-1-2.
FLAVONOID DETERMINATION 22
The plant sample (1 g) was extracted repeatedly with aqueous methanol (20 ml in 80%) (CH3OH) at room temperature. The whole solution was filtered through whatman filter paper No 42 (125 mm). The filtrate was later transferred into a crucible and evaporated into dryness over a water bath ansd weighed to a constant weight. The results are given in Table.
SAPONIN DETERMINATION 23
Each of the powder samples (5 g) were put into a conical flask and 20% aqueous ethanol (25 ml) was added. The samples were heated over a hot water bath for 4 hours with continuous stirring at about 55oC. The mixture was filtered and the residue re-extracted with 20% aqueous ethanol (50 ml). The combined extracts were reduced to 10 ml over water bath at about 90oC. The concentrated extract (was transferred into a separatory funnel and diethyl ether (5 ml) was added and shaken vigorously. The aqueous layer was recovered while the whether layer discarded. The purification process was repeated. n-butanol (15 ml) wad added and n-butanol extracts were washed twice with aqueous 5% NaCl (3 ml of) solution). The remaining solution was heated on a water bath. After evaporation the samples were dried in an oven to a constant weight; the saponin content calculated as percentage. The results are given in Table.
Table-2.2: Amount (% on dry powder basis) of crude alkaloid, flavonoid and saponin on the medicinal plants investigated.
Plants Alkaloids (%) Flavonoids (%) Saponin (%)
Polygonum plebejum 0.8 14.06 8.5
2.2.2 EXTRACTION AND ISOLATION OF COMPOUNDS FROM POLYGONUM PLEBEJUM
Plant powder was taken in a few precleaned cloth thimbles. The thimbles containing the powder were placed in a Soxhlet apparatus.
The plant powder extracted separately and exhaustively in Soxhlet apparatus first with Petroleum ether (boiling point 40-60 oC and 60-80 oC) followed by ethyl acetate. All the extracts were filtered individually; the filtrate was combined together and then concentrated in a “Buchi Rotavapor” under reduced pressure. This was shown in scheme-1.
2.2.2.1 EXTRACTION SHEME OF POLYGONUM PLEBEJUM
Powder (~950g)
Extraction with pet-ether (b.p.40-6oC)
Petroleum ether exract Residue
Evaporated to dryness Extracted with Ethyl acetate
Gummy mass Ethyl acetate extract Residue
(~20 g) Evaporated to dryness
Dry mass of ethyl acetate extract (~18 g)
Scheme 1: Extraction scheme of Polygonum plebejum
2.2.3 INVESTTIGATION OF ETHYLACETATE EXTRACT
2.2.3.1 THIN LAYER CHROMATOGRAPHY (TLC) STUDY
TLC analysis of the ethylacetate extract showed the presence of one spot (Petroleum ether: Dichloromethane = 90: 20) having light brown under iodine vapor. Furthermore, TLC study showed the presence of two spots on spraying with vanillin sulfuric acid followed by heating in oven for 15 minutes. Again three spots (Dichloromethane: Ethyl acetate = 80: 20) were visible on spraying vanillin sulfuric acid followed by heating in an oven for 15 minutes. Among these three spots, two were pink and one was violet. The presence of the pink colored spot was thought to be due to the presence of steroidal or fatty acid material or both of them. It gave also positive test for steroid23.
2.2.3.2 FRACTIONATED BY VACCUM LIQUID CHROMATOGRAPHY (VLC)
The ethyl acetate extract was concentrated to dry mass about 18g by a ‘Buchi Rotavapor’. The concentrated extract was mixed with TLC grade silica gel (60 GF254). The mixture was made as powder for using in column. Then the sample was placed on the top of the bed of silica gel packed column (VLC). The column was then eluted with petroleum ether followed by mixtures of pet-ether and dichloromethane of increasing polarity. The elutes were collected in test tube with an about of about 20mL in each. Solvent system used as mobile phase in the analysis of ethyl acetate extract was listed in Table.
2.2.3.3 SCREENING OF THE FRACTIONS
2.2.3.4 ATTEMPT OF PURIFICATION AND CHARACTERIZATION OF THE FRACTIONS
2.2.3.4.1 ANALYSIS OF FRACTION F1
The fraction F1 was light yellow in colour. This was left undisturbed at room temperature for several days. It gave no crystals and gave an oily appearance. Then, the TLC study of the sample showed one spots on TLC plates on spraying with vanillin sulfuric acid followed by heating in an oven for 10 minutes. It was not studied further because no signal in Infra-Red Spectrum was observed.
2.2.3.4.2 ANALYSIS OF FRACTION F2
The fraction F2 was yellowish in colour. The fraction was left undisturbed at room temperature for several days. It gave white crystals at the bottom of the test tube. The TLC study of this showed one spot of pink colour on spraying with vanillin sulfuric acid followed by heating in oven at 110oC for 10 minutes.
2.2.3.4.3 ANALYSIS OF FRACTION F3
The fraction F3 left undisturbed at room temperature for several days but no crystal was obtained. This was concentrated to a deep colored semisolid. Its TLC study showed two violet spots with tailing by developing vanillin sulfuric acid spray.
2.2.3.4.4 ANALYSIS OF FRACTION F4
The fraction F4 left undisturbed at room temperature for several days. No crystal formed in this fraction and no distinct spot was observed in TLC study.
2.2.3.4.5 ANALYSIS OF FRACTION F5
The fraction F5 was concentrated to a small volume and was left undisturbed at room temperature. After several days a semisolid substance was obtained. It was soluble in hexane. It’s TLC study did not give any information result.
2.2.3.4.6 ANALYSIS OF FRACTION F6
The fraction F6 was concentrated to a small volume and was left undisturbed at room temperature. After sever days an solid substance was obtained. It was washed with hexane very well. After washing, the solid was dissolved in chloroform and the solution was kept in a vial for recrystallization at room temperature and crystals were obtained. Its TLC study also showed a single spot in solvent system (Petroleum ether: Dichloromethane = 20:80). The spot was pink and turned into violet colour by spraying with vanillin sulfuric acid. It was studied further.
2.2.3.4.7 ANALYSIS OF FRACTION OF F7
The fraction F7 was treated with charcoal to absorb chlorophyll. After treating, the fraction was filtered and left undisturbed at room temperature for several days. But no crystal was found.
2.2.3.4.8 ANALYSIS OF FRACTION F8
The fraction F8was treated with charcoal to absorb chlorophyll. After treating, the fraction was filtered and left undisturbed at room temperature for several days. But no crystal was found.
2.2.3.4.9 PROPERTIES OF THE ISOLATED COMPOUNDS
Properties of S.P-1
Physical state: Solid.
Solubility: Soluble in chloroform and hexane.
Rf value: 0.43 (over silica gel, GF254, petroleumether: dichloromethane = 90:10 as the mobile phase).
Amount: 0.892 mg.
Spectral characteristics:
The compound S.P-1 had important frequencies of absorption at 3400, 2850, 2900, 1720, 1650, 1350, 1240, 900, 800 cm-1.
Properties of SPPA1:
Physical state: Light yellow crystal.
Solubility: Soluble in chloroform & methanol.
Rf value: 0.5 (over silica gelGF254, petroleumether: dichloromethane
=20:80 as the mobile phase).
Melting point: 59-61oC
Amount: 4.106 mg.
Spectral characteristics:
The compound SPPA1 had important frequencies of absorption at 3400, 2850, 1700, 1640, 1475,1440, 1370, 1240, 740, 720 cm-1.
The compound SPPA1 had important chemical shift at 1.67(S), 2.006(d, J=6.6 Hz), 2.16 (s), 2.3286(t), 4.08(d), 5.3327(m), 0.8503(m), 1.5(m), 1.6016(m), 1.2446(board m), 2.3472(t) ppm.
The compound SPPA1 had important chemical shift value at 19.166, 31.937, 29.529, 31.937, 33.967,173, 179, 39.8, 24.720, 64, 118, 142, 22,87, 61 ppm.
Properties of SP2:
Physical state: Light yellow oily substance.
Solubility: Soluble in petroleum ether, ethyl acetate,chloroform & methanol.
Amount: 6 mg.
Spectral characteristics:
The compound SP2 had important frequencies of absorption at 3030, 2900, 2850,1735,1450,1370,1260,1030 800 cm-1.
Properties of SP3:
Physical state: Greenish gummy material.
Solubility: Soluble in petroleum ether, ethyl acetate, chloroform & methanol.
Amount: 26 mg.
Spectral characteristics:
The compound SP3 had important frequencies of absorption at 3400,2905, 2870, 1720, 1640, 1450,1375,1060,1020,800cm-1.
2.3 Result and discusion
The spcies Polygonum plebejum (bishkatali) of the family polygonaceae has been collected locally, dried, powdered and extracted with petroleum ether followed by ethylacetate. Ethyl acetate extract of this species was fractionated by VLC (vacuum liquid chromatography) & different fractions obtained were separated & purified using different chromatographic techniques. The compounds isolated from this extract were characterized by different physical and spectroscopic studies.
3.1 CHARACTERISATION OF COMPOUNDS ISOLATED FROM ETHYL ACETATE EXTRACT OF POLYGONUM PLEBEJUM
3.1.1 CHARACTERISATION OF COMPOUND S.P-1
The compounds S.P-1 (0.892 mg) was isolated as white crystalline powder from the ethyl acetate extract of the plant Polygonum plebejum. It was soluble in ethyl acetate. TLC examination of S.P-1 indicated that it was a single compound (Rf value = 0.43 (over silica gel, GF254, petroleum ether: dichloromethane = 90:10 as the mobile phase). It was visualized as a brownish coloured spot upon its exposure to iodine vapour and as a violet coloured spot on sparaying with vanillin-sulfuric acid followed by heating in an oven at 110oC.
IR SPECTROSCOPIC STUDY
The band at 2900 cm-1 & 2850cm-1 were due to the presence of aliphatic C-H stretching of either both –CH3, -CH2- or >CH- group24. The peak at 1640 cm-1 was suggestive the presence of >C=C< group. The peak at 1450 cm-1 was due to the presence of –CH2- group. The absorption band at 1720 cm-1 was suggestive of C=O stretching for normal aliphatic ester. The peak at 1240 cm-1 indicated about C-N stretching. The peak at 1550cm-1 indicated that primary & secondary amines (bending) might be present. The peak at 900 cm-1 for aromatic stretching (out of plane bend). And the peaks at 830 cm-1 & 800 cm-1 was indicated the presence of >C=C-H.
3.1.2 CHARACTERISATION OF THE COMPOUND SPPA1
The compound SPPA1 (4. 106 mg) was isolated as light yellow crystal from the concentrated ethyl acetate extract of the plant Polygonum plebejum upon vacuum liquid chromatography and crystallization of the fraction F6. It was soluble in chloroform and methanol. Its TLC study indicated the presence of a single spot (Rf= 0.5) in over silica gelGF254, petroleum ether: dichloromethane =20:80 as the mobile phase). The spot was visualized as a violet colou upon sparaying with vanillin-sulfuric acid followed by heating in an oven at 110oC.
IR SPECTROSCOPIC STUDY
The IR spectrum30 (Fig-2.4 ) showed an absorption band at (3400-3300) cm-1 due to O-H stretching. The band at (3000-28500 cm-1 was for C-H stretching. The band at 1700 cm-1 due to C=O stretching. The band at 1450 cm-1 & 1370 cm-1 were indicated that methylene group & methyl group have a characteristic bending absorption. The absorption at (1200-1000) cm-1 was for C-O single bond stretching. The other band at 1640 cm-1 for >C=C< stretching and (900-740) cm-1 was due to C-H (out of plane) aromatic stretching. The other band 720 cm-1 was indicated the presence of long chain band. 1H-NMR SPECTROSCOPY The 1H-NMR31 (400MHz in CDCl3) spectrum (Fig-2.5) of the isolated compound SPPA1 had two sharp singlet at 1.67 and 2.16 ppm indicative of methyl protons at C-4 and acetyl at C-3 respectively32. A doublet at 4.08 ppm with coupling constant, J=6.72 Hz indicative the presence of oxymethine proton at C-6. The spectrum also showed a triplet at 2.3286 ppm exhivitive to the presence of oxymethine proton at C-2. The distorted multiplet at 5.3327 ppm was indicative the presence of an olefinic proton33 at C-5. 1H-NMR spectrum of SPPA1 suggested that the structure had a isostearate group attached to C-1. 1H-NMR spectrum showed one multiplet at 0.8503 ppm for two terminal methyl groups in C-17’& C-18’. The singal (multiplet) of the next methane proton was observed at 1.6016 ppm at C-3’. The broad and intense singal at δ 1.2446 ppm (C-4’ to C-15’) appeared as a triplet at δ 2.3472 ppm (C-2’). The assignment of the proton singals has been compared with literature value of isostearic acid34. Table-3.1: 1H-NMR spectral data of compound SPPA1 Protons Chemical Shift value of the isolated compound SPPA1 in ppm Assignmnt Methyl proton 1.67 Singlet Methine proton at C-3 2.006 Doublet (J=.6 Hz) Acetyl methyl proton 2.16 Singlet Methine proton at C-2 2.3286 Triplet Methine proton at C-1 4.08 Doublet Olefinic proton at C-5 5.3327 Multiplet Two methyl proton at C-17’, C-18’ 0.8503 Multiplet One methane proton at C-16’ 1.5 Multiplet Methylene proton at C-3’ 1.6016 Multiplet Methylene proton at C-4’ to C-15’ 1.2446 Broad multiplet Methylene proton at C-2’ 2.3472 Triplet 13C-NMR SPECTROSCOPY The 13C-NMR spectrum (Fig-2.9) of the isolated compound SPPA1 revealed the presence of 27 carbon signals. The signal at chemical shift,δ 24.720 (C-4) was assigned to a methyl carbon. The signals at 61 (C-1), 87(C-2), 22(C-3) and 64(C-6) ppm were assigned to four methane carbons respectively35. The signal at 142 (C-4) and 118(C-5) ppm were assigned for the >C=C< carbons35. The signal at 179 ppm was assigned for acetyl carbonyl carbon attached to C-3 and 39.8 ppm was assigned for acetyl methyl carbon. 13C spectrum of SPPA1 also suggested that the structure had an isostearate group. The signal at chemical shift at 173 (C-1’) ppm was attributed to ester carbon. The signal at δ 33.967 ppm (C-2’), 31.937 (C-3’) and 29.529 (C-4’ to C-15’) were assigned to methylene carbons. The signal at δ 19.166 (C-17’, C-18’) ppm were due to the presence of terminal methyl carbon of isopropyl group and signal at 31.937 (C-16’) ppm suggested the presence of methane carbon of isopropyl group. The assignment of the carbon signals has been compared with the literature value of isostearic acid35. Table-3.2: 13C-NMR spectral data of the isolated compound SPPA1 No. of carbon Types of carbon (assigned) Chemical shift value (Observed) C-1 CH 61 C-2 CH 87 C-3 CH 22 C-4 Olefinic 142 C-5 Olefinic 118 C-6 CH 64 Methyl carbon at C-4 CH3 24.720 Acetyl methyl carbon at C-3 Acetyl methyl 39.8 Acetyl carbonyl carbon at C-3 Acetyl carbonyl 179 C-1’ Ester carbon 173 C-2’ Methylene carbon 33.967 C-3’ Methylene carbon 31.937 C-4’ to C-15’ Methyne carbon 29.529 C-16’ Methylene carbon 31.937 C-17’ to C-18’ Methyl carbon 19.166 Combining IR, 1H-NMR and 13C-NMR & DEPT NMR spectroscopic data, the compound SPPA1 was identified as 2,6-dihydroxy-3-acetyl-4-methyl-cyclohex-4-enyl isostearate having the structure as given below: It appears from literature survey that the isolation of this compound from this plant had been reported. All these spectral data36 were in good agreement with the reported spectral data. The reported compound SPPA1 possessed some impurity like benzene containing ring, so a few unwanted signal was appeared. Because of lake of time, this cannot be further investigating. This compound is a known compound, isolated from petroleum ether extract, but it was isolated for the first time from the ethyl acetate extract of Polygonum plebejum. 3.1.2 CHARACTERISATION OF THE COMPOUND SP2 The compound SP2 (6 mg) was isolated as light yellow oily substance from the concentrated ethyl acetate extract of the plant Polygonum plebejum upon vacuum liquid chromatography and crystallization of the fraction F3. it was soluble in chloroform and methanol. It gives mixture compounds & due to lack of time it could not be analyzed extensively. However, spectral (Fig-1.14) work is in progress. IR SPECTROSCOPIC STUDY The IR spectrum30 (Fig-2.14 ) showed a tiny absorption band at 3030 cm-1 indicative the unsaturation of the compound. Another strong absorption band at 800 cm-1 indicated the presence of CH out of plane deformation.. The band at 2900 cm-1 and 1370 cm-1 were indicative of methyl groups while the bands at 2850 cm-1 and 1450 cm-1 were diagnostic of –CH2- groups. An intense absorption band at 1735 cm-1 which indicated the presence of an ester group. This argument was supported by the presence of bands at 1260 cm-1, 1100cm-1, 1030 cm-1 for C-H stretching. From the above discussion, it was evident that the compound SP2 was either an unsaturated fatty acid or a lactone. 3.1.2 CHARACTERISATION OF THE COMPOUND SP3 The compound SP3 was isolated as greenish gummy material from the concentrated ethyl acetate extract of the plant Polygonum plebejum upon vacuum liquid chromatography and crystallization of the fraction F7. It was soluble in petroleum ether. Due to lack of time it could not be analyzed extensively. However, spectral work was in progress. IR SPECTROSCOPIC STUDY The IR spectrum (Fig-2.15) showed an absorption band at 3400cm-1 due to hydrogen bonded O-H stretching. It also showed carbonyl stretching at 1720 cm-1. The presence of carbonyl group was supported by the presence of C-O stretching at 1060 cm-1 and 1020 cm-1. The band at 2905 cm-1 & 1375 cm-1 were indicated that methyl group has a characteristic bending absorption. The absorption at 2870 cm-1 and 1450 cm-1 were due to the presence of –CH2- group. The other band at 1640 cm-1 for >C=C< stretching.
From the above discussion, it was evident that the compound SP3 was a mixture of unsaturated fatty acids.
3.1 FATTY ACID ANALYSIS
3.1.1 INTRODUCTION
The fatty acid series was so named because some of the higher members, particularly palmitic and stearic acids occur in natural fat24. Fats are glycerides of fatty acids. Since glycerol is a trihydroxy alcohol and fatty acids are monobasic, a normal glyceride is a triglyceride and on hydrolysis yields three molecules of fatty acids and molecules of glycerol.
C3H5(C18H35O2)3 + 3H2O = C3H5(OH)3 + 3C18H36O2
The great numbers of the naturally occurring fatty acid belong to a few homologus series. The general formula of the fatty acids is CnH2nO2 may represent the series to which stearic acid belongs. As, however, their functional group is the carboxyl group, COOH, they are more conveniently expressed as CnH2nCOOH, since these show the nature of the funtional group. Nearly all the naturally occurring fatty acids have even number of carbon atoms in the molecule. The members of greatest importance are shown in Table-2.1.
Table- 3.1: SOME COMMON FATTY ACIDS FOUND IN PLANT AND ANIMAL KINGDOM.
Common name Carbon
atoms Double
bonds IUPAC name Source
Butyric acid 4 0 Butanoic acid Butter fat
Caproic acid 6 0 Hexanoic acid Butter fat
Caprylic acid 8 0 Octanoic acid Coconut oil
Capric acid 10 0 Decanoic acid Coconut oil
Lauric acid 12 0 Dodecanoic acid Coconut oil
Myristic acid 14 0 Tetradecanoic acid Plam Kernel oil
Palmitic acid 16 0 Hexadecanoic acid Palm oil
Palmitoleic acid 16 1 9-hexadecenoic acid Animal Fats
Stearic acid 18 0 Octadecanoic acid Animal fats
Oleic acid 18 1 9-octadecenoic acid Olive oil
Vaccenic acid 18 1 11-octadecenoic acid Butter fat
Linoleic acid 18 2 9,12-octadecadienoic acid Grape seed oil
Alpha-linoleic acid (ALA) 18 3 9,12,15-octadecatrenoic acid Flax seed
Gamma-linolenic acid (GLA) 18 3 6,9,12-octadecatrienoic acid Borage oil
Arachidic acid 20 0 Eicosanoic acid Peanut oil, fish oil
Gadoleic acid 20 1 9-eicosanoic acid Fish oil
Arachidonic acid 20 4 5,8,11,14-eicosatetraenoic acid Liver fats
EPA 20 5 5,8,11,14,17-eicosapentaenoic acid Fish oil
Behenic acid 22 0 Docosanoic acid Rapeseed oil
Erucic acid 22 1 13-docosenoic acid Rapeseed oil
DHA 22 6 4,7,10,13,16,19-docosahexaenoic acid Fish oil
Lignoceric acid 24 0 Tetracosanoic acid Small amounts in most fats
The fatty acids in plants are generally straight-chain compounds, ranging from three to eighteen carbons. Fatty acids containing an even number of carbon atoms are present in substantial amount because they are built up of two carbon atoms at a time, from acetic acid units. Fatty acids may be saturated or unsaturated; the saturated acids can also exist in both cis- and trans- forms.
Fatty acids isolable in water and are extracted from plant tissues by organic solvents of low polarity like petroleum ether or chloroform. They are generally found in ester linkage but are also found free and in loose molecular complexes with protein. Although alkaloids, terpenoids, steroids, flavones and their glycosides, phenols, and phenolic acids constitute a major portion of non-carbohydrate materials of a plant, fatty acids are always present in varying amounts in all plant materials.
These fatty materials may influence the handling of the plant tissues as well as any chemical treatment done on it. Therefore, the study of fatty acids, the major constitute of all fatty matters is important.
Fatty acids occur in plants in bound form25 as fats or lipids. Fats are the triglycerides of fatty acids of the same type or of the different types and yield fatty acids upon hydrolysis. Lipids are defined by their special solubility properties and are comprised of different kinds of compounds. These lipids comprise up to 7% 25of the dry weight in leaves in higher plants, about 1-15% in stems of green plant 17 and are important as membrane constitutes in the chloroplasts and mitochondria lipids also occur in considerable amounts in the seeds or fruits of a number of plants. Although numerous fatty acids are now known in plants the palmitic acid is the major saturated acid 25 in leaf lipids and also occur in varying quantities in some seeds oils. Stearic acid is the major saturated acid in seed fats of a number of plant families 25. Unsaturated acids (mainly C16 and C18) are widespread in both leaf and seed oils. A number of rear fatty acids 25 (e.g., erueic and sterculic acid) are found in seed oils of a few plants. Seed oils from plants such as olive, plam, coconut etc. are exploited commercially and are used as edible oils, for soap manufacture and in the pint industry. Fats are sometimes converted into the methyl esters of carboxylic acids by the reaction with methanol in presence of a catalytically reduced to straight-chain primary alcohols of even carbon number, and from these a host of compounds can be synthesized.
3.1.2 GAS CHROMATOGRAPHY (GC)
Chromatography26 is a physical process of separation in which the components to be separated are distributed between two phases a stationary phase having large surface area (for example a porous solid) and a mobile phase (a gas or liquid) moving in contact with the stationary phase which is generally coated on a porous solid phase. Different forms of chromatography are named according to the physical state of mobile phase employed; further subdivision is done according to the type of interaction between the solute and stationary phase. Now each type of chromatography can be subjected to three forms of developments; Elution, Frontal analysis or Displacement. In elution technique a carrier fluid (eluent) which is relatively invert towards the stationary phase, is kept flowing continuously and components of sample, though mutually resolved, are in mixture with the carrier fluid. Actually it is this technique that is generally employed in gas chromatography (GC) is fundamentally a separation technique but it provides identification of a compound and with due calibration permits quantitative estimation as well. Today it is used by analytical chemists in every branch of science as a powerful analytical tool. It finds application in various fields such as gases and pollutants, petroleum products, oils and fats, foods and flavours, drugs, beverages, elemental organic analysis and a number of varied materials.
3.1.3 THE PRINCIPLE ADVENTAGES OF GC
The principal advantages of GC to an analyst are
i. The technique has strong separation power and even quite complex mixtures can be resolved into constituents.
ii. It is a micro method and only a few mg samples are enough for analysis; sensitivity of the method is very high.
iii. Speed of analysis is quite fast.
iv. Gives good precision and accuracy.
v. It involves relatively simple instrumentation; operation of a gas chromatograph and related calculation do not require highly skilled personnel and thus the technique is very suitable for routine analysis.
vi. The cost of equipment is relatively low and life is generally long.
Thus in large number of instances GC is employed as the only method able to provide the desired results, in other cases it forms better alternative test method.
Any sample that can be vaporized (or the components could assume a vapor pressure of at least few mm of Hg) without thermal decomposition at the operating temperature could be analyzed by GC. At present due to various reasons, including difficult instrumentation and lack of high temperatures materials, the separating temperature is generally limited to about 4500C. Samples that cannot be vaporized are converted into volatile derivatives (for examples, fatty acids converted into methyl esters) and subjected to GC analysis.
3.1.4 PRINCIPLE OF GC SEPERATIONS
When some gas or vapor comes in contact with an absorbent, a certain amount of it gets absorbed on the solid surface; according to the well known laws of Fraundilich (x/m = KC1/n), x is the mass of the vapour adsorvent in mass ‘m’ of (dissolved) in the bulk liquid; which follows the partition law of Henry (x/m = kC). Now both the sorption phenomena are selective and there are different k-values (distribution co-efficient), in general, for different vapor sorbent pairs; for the same sorbent, different vapors will have different K-values, i.e. varying affinities.
Gas chromatographic separation is accomplished in a tubular column made of glass, metal or Teflon. The column is filled with a sorbent as the stationary phase, absorbents are packed as such in the from of fine size graded powder whereas liquids are either coated as fine film on the column wall of first coated over an inert porous support such as fine powder and then packed into the column. A gas such as hydrogen, nitrogen, helium etc. serving as mobile phase, flows continuously through the column. It is called the carrier gas and serves to transport the sample components in the column. The sample is introduced as a sharp plug of vapour at the carrier gas entrance end of the column. Different components of the sample are absorbed on the stationary immediately there after, the portion of each component in the gas phase is swept further by the carrier gas and so a fraction of the sorbed amount also desorbs out to maintain the k-value; at the same time, out of the swept amount some portion will go into sorbent at the next point in the column, again to maintain the k-value. This goes on successfully and continuously and as a sharp of, more or less Gaussian Distribution (peak shape). Now components having varying affinities for the sorbent (stationary phase) will held up in the latter to different degrees and consequently will move along the column with different speeds; if a column of sufficient length is chosen, these components will emerge out of the column at different intervals. This is how the separation of components is achieved in GC.
3.1.5 PROCEDURE OF FATTY ACID ANALYSIS 27
2.1.5.1 ANALYSIS OF FATTY ACIDS
Petroleum ether extract (0.2731g) of Polygonum plebejum was dissolved in petroleum ether (100ml) and extracted with 5% sodium bicarbonate solution (25mL X 2). The mixture was taken in a separatory funnel and shaken vigorously and allowed to stand for overnight. Two layers were obtained. The lower layer (aqueous) was separated and taken for the analysis of free fatty acid (FFA). The upper layer was separated and taken for the analysis of bound fatty acid (BFA).
3.1.5.2 ISOLATION OF FREE FATTY ACID (FFA)
The lower part was acidified (pH 2.5) by 2M sulphuric acid. The mixture was then extracted with petroleum ether (b.p-40-600) (25mL X 3). The pet ether fraction was dried over anhydrous sodium sulphate, filtered and the filtrate was evaporated to dryness.
Now the saponified materials obtained from the pet-ether extract was taken in a pear shaped flask and 2 mL of borontrifluride-methanol (BF3-MeOH) complex was added and the mixture was refluxed on a boiling water bath for30 min. The mixture was then evaporated in a rotavapor to dryness and transferred in a small separatory funnel containing a little water (6mL). The mixture was shaken vigorously and then extracted with hexane. The aqueous layer was discarded. The hexane part containing the methyl esters of fatty acids was made free from water by adding anhydrous sodium sulphate. The solution was filtered and the filtrate was concentrated for the analysis of free acids by GLC (Shimadzu 9A, Column-BP-50, Detector- FID, 1700C-1 min/40C-2700C-30min).
3.1.5.2.1 ISOLATION SCHEME FOR FATTY ACIDS FROM POLYGONUM PLEBEJUM
Petroleum ether exact of Polygonum plebejum
Add 5% of NaHCO3
Mixture (shaken vigorously)
Transferred in separatory funnel
Separate
Organic later Aqeous part
(Unreacted fatty material)
BFA
Add 10mL of 0.05 M NaOH solution
& shaken
Reflux 30 minutes
Na-salt of FFA + Glycerol
Evaporated by rotavapour Transferred in a separatory funnel
+ Add distilled water + add Hexane & shake
Taken Hexane part
Aqueous part is taken + add 2M H2SO4
& transferred in a separatory funnel in a beaker
Add Na2SO4
& filter
Filtrate
(Dryness & weighed which is total BFA)
Add 1 mg of Benzoic acid & 2mL BF3-MeOH complex
Reflux 20 minutes & evaporated
Transferred in separatory funnel & 6mL-distilled water
Add hexane & shaken, taken hexane part
Add Na2SO4 & filter
Concentrated & transferred into a vial
Send for GLC
2nd part
Aqueous part (Na-salt of fatty acid)
2M H2SO4 added to
control pH 2.5
Pet ether is added and shak
Transferred in a separatory funnel
& taken Pet-ether part into a beaker
Anhydrous Na2SO4 was added & filter
Weighed pear shaped flask,
transferred filtrate to it and dryness by rotavapour
Again weighed which is Total FFA
Add 1 mg of C6H5COOH & 2mL of BF3-MeOH complex
Reflux on boiling water bath, minimum 30 minutes
Dryness by rotavapour & transferred in a separatory funnel
Add 6mL of distilled water,
firstly shaken and add hexane
Taken hexane part in a beaker which is FFA of methyl ester
Add anhydrous Na2SO4, filter & concentrated by rotavapour
Transferred in a vial
Send for GLC
Scheme: 2 Fatty acid analysis
3.1.5.3 ISOLATION OF BOUND FATTY ACID (BFA)
The upper part was taken in appear shaped flask and was added 10 mL of 0.5 M NaOH solution, was shaken well. The mixture was refluxed for 30 min in a boiling water bath. Then the mixture was evaporated to dryness by means of a rotavapour. A little water was added to the mixture and transferred to a seperatory funnel to settle down. The non-saponified materials were separated from the saponified portion (aqueous layer) by extraction with hexane.
The aqueous layer containing fatty acids (as salts) was acidified by adding 2M sulphuric acid and to pH 2.5. The mixture was extracted with hexane. The hexane part was taken in a conical flask and made from water free by adding anhydrous sodium sulphate and then filtered. The filtrate contained saponified materials.
Now the saponified materials obtained from the pet ether extract was taken in a pear shaped flask and 2 mL of borontrifluride-methanol (BF3-MeOH) complex was added and the mixture was refluxed on a boiling water bath for 3m min. The mixture was then evaporated in a rotavapor to dryness and transferred in a small separatory funnel containing a little water (6mL). The mixture was shaken vigorously and then extracted with hexane. The aqueous layer was discarded. The hexane part containing the methyl esters of fatty acids was made free from water by adding anhydrous sodium sulphate. The solution was filtered and the filtrate was concentrated for the analysis of bound fatty acids by GLC (Shimadzu 9A, Column-BP-50, Detector-FID, 1700C-1 min/40C-2700C-30min).
3.1.6 RESULT
TABLE-3.2: STANDARD RETENTION TIME (RT) OF DIFFERENT METHYL ESTERS OF DIFFERENT FATTY ACIDS FROM GC CHROMATOGRAM.
Standard
RT (min) Fatty acid
1.16 Caprylic acid
3.07 Capric acid
6.15 Lauric acid
9.50 Myristic acid
12.27 Palmitoleic acid
12.72 Palmitic acid
12.72 Oleic acid
15.23 Stearic acid
15.64 Arachidic acid
18.31 Behenic acid
20.80 Lignoceric acid
TABLE-3.3: BOUND FATTY ACIDS IN PETROLEUM ETHER EXTRACT OF POLYGONUM PLEBEJUM
RT (min) Fatty acid
15.37 Stearic acid
12.74 Oleic acid
9.46 Myristic acid
6.50 Lauric acid
20.48 Lignoceric acid
Free fatty acids in petroleum ether extract of Polygonum Plebejum was not found here.
3.1.7 Discussion
Fat is important for many body processes and some fat are needed to eat in daily diet. Dietary fats are classified by their structure. Different types of fats react differently inside the body. Saturated fats (found mostly in animal products) increase blood cholesterol, which is a risk factor in coronary heart diseases. Unsaturated fats (found mostly in plant and marine foods) tend to lower blood cholesterol, The composition analysis of petroleum ether extract of Polygonum plebegum by GC revealed that the bound fatty acid portion of this plant was rich in unsaturated fatty acids (such as). The plant Polygonum plebejum contain small amount of fatty acids. Thus these plants are very important for edible purpose and local people used to taken this plant as leafy vegetables.Further analysis is necessary to know more about the content of fatty acids of this plant.
3.2 ASH CONTENT ANALYSIS
3.2.1 ASH:
The ash content is the percentage of inorganic residue remaining after ignition of sample.
3.2.2 PROCEDURE:
A weighed quantity of sample was taken in a crucible. The contents were heated carefully to the ignition point and allowed to burn spontaneously. When the burning was completed, the crucible heated at a dull-red heat in a muffle-furnace at 600ºC for three hours. The crucible was then cooled and weighed with accuracy28.
3.2.3 CALCULATION:
100 X W2
Ash percentage = ───────
W1
Where, W1 = Initial weight of the sample in g.
W2 = Weight of the ash in g.
3.2.4 RESULT:
The result of the determination of the amount of ash content of the plant powder of Polygonum plebejum been calculated and given in the following table.
TABLE-3.4: THE ASH CONTENT OF THE PLANT POWDER OF POLYGONUM PLEBEJUM.
Sample
Weight of crucible (g)
Weight of sample taken in the crucible W1 (g) Weight of crucible + ash (g)
Weight of ash obtained W2 (g)
Percentage
(%)
Powder of Polygonum plebejum 28.4735 2.0018 28.4753 0.2981 14.89
3.3 MOISTURE ANALYSIS
MOISTURE
3.3.1 PROCEDURE
The plant powder was taken in a weighing bottle and placed in a oven at100 oC for several days .The weighing bottle containing sample was weighed after cooling at room temperature in dessicator. Again it was placed in oven and weighed after cooling in several times. When a constant weight was found, it was recorded.
3.3.2 RESULT
The result of the determination of moisture content of the plant powder of Polygonum plebejum is given in table.
TABLE-3.4: THE MOISTURE CONTENT OF THE PLANT POWDER OF POLYGONUM PLEBEJUM
Sample Weight of weighing bottle (g) Weight of sample taken in the weighing bottle, W1 (g) Weight of weighing bottle after loosing moisture (g) Weight of moisture obtained W2 (g) Percentage (%)
Plant powder of Polygonum plebejum 19.80 (24.7506-19.80)
= 4.9506 24. 0553 4.2553 14.04
4. MICROBIAL ACYIVITY
4.1 INTRODUCTION:
Bacteria and fungi are responsible for many infectious diseases. The increasing clinical implications of drug resistant fungal and bacterial pathogens have lent additional urgency to antimicrobial drug research. The antimicrobial screening that is the first stage of antimicrobial drug research is performed to ascertain the susceptibility of various fungi and bacteria to any agent. This test measures the ability of each test sample to inhibit the in vitro fungal and bacterial growth. Anti-microbial activity of herb extracts was evaluated with antibiotic susceptible and resistant microorganisms. Many herbs have been used because of their antimicrobial trait, which are due to compounds synthesized in the secondary metabolism of herb. Their active substances, for example, phenolic compounds that are part of essential oils.
This ability may be estimated by any of the following three methods.
Disc diffusion method
Serial dilution method
Bioautographic method
Among the above-mentioned techniques the disc diffusion (Bauer et al., 1966) is a widely accepted in vitro investigation for preliminary screening of test agents that may possess antimicrobial activity. It is essentially a quantitative or qualitative test indicating the sensitivity or resistance of the microorganisms to the test materials.
4.2 PRINCIPLE OF DISC DIFFUSION METHOD 29
Solutions of known concentration (μg/mL) of the test samples are made by dissolving measured amount of the samples in calculated volume of solvents. Dried and sterilized filter paper discs (6 mm diameter) are then impregnated with known amounts of the test substances using micropipette. Discs containing the test material are placed on nutrient agar medium uniformly seeded with the test microorganisms. Standard antibiotic discs and blank discs (impregnated with solvents) are used as positive and negative control. These plants are then kept at low temperature (4 oC) for 24 hours to allow maximum diffusion. During this time dried discs absorb water from the surrounding media and then the test materials are dissolved and diffused out of the sample disc. The diffusion occurs according to the physical law that controls the diffusion of molecules through agar gel. As a result there is a gradual change of test materials concentration in the media surrounding the discs.
These plates are then incubated at 37º C for 24 hours to allow growth of the organisms. If the test material has any anti-microbacterial activity, it will inhibit the growth of the microorganisms and a clear, distinct zone of inhibition in mm will be visualized surrounding the medium. The antimicrobial activity of the test agent is determined by measuring the diameter of zone of inhibition expressed in millimeter.
The experiment is carried out more than once and the mean of the mean of the readings is required.
4.3 EXPERIMENTAL
4.3.1 TEST MATERIALS
The herbs were collected and separated from unwanted materials. The herbs were washed with water to remove dust, mud and then dried at 50ºC overnight. Dried herbs were then ground to powder in a grinding machine.
4.3.2 EXTRACTION
Dry powders of the herbs were extracted successively with chloroform, ethyl acetate, methanol and water.
4.3.2.1. EXTRACTION OF THE COMPONENTS SOLUBLE IN CHLOROFORM
The powder (10 g) was taken in a conical flask(150ml). Chloroform (40 ml) was added to the powder and was kept at 25ºC for 12 hours. The suspension was filtered successively through a piece of sterile cotton fabric and a filter paper (Whatman). The filtrate was then evaporated by vacuum dryer at 40ºC overnight to get the chloroform extract.
4.3.2.2 EXTRACTION OF THE COMPONENTS SOLUBLE IN ETHYL ACETATE
The solid residue left after chloroform extract was dried at 40ºC overnight to remove residual chloroform. The solid residual powder was added to ethyl acetate (40ml) and kept at 25ºC for 12 hours, and ethyl acetate extract was recovered by the same procedure like chloroform extract.
4.3.2.3. EXTRACTION OF THE COMPONENTS SOLUBLE IN METHANOL AND WATER
The solid residue left after ethyl acetate extract was dried at 40ºC overnight to remove residual ethyl acetate. The solid residual powder was added to 75% methanol (40 ml) and kept at 25ºC for 12 hours. The suspension was separated successively through centrifugation at 12000 rpm, for 5 minutes. The filtrate was then dried overnight in a beaker at 50ºC to get the (75%) methanol extract. The solid residue was dried at 40ºC overnight to remove residual methanol (75%). Vacuum dryer at 40ºC then evaporated the filtrate for 6 hours to get the methanol and water extract. Then 40ml-distilled water was added to the powder containing methanol and water extracts and suspension was collected. Then centrifugation was done again at 12000 rpm for 5 minutes, filtrate containing water-soluble compounds was collected, dried in vacuum dryer at 40ºC for 6 hours and finally the powder containing water soluble part was obtained as the water extract. The solid residue was dried in a vacuum dryer, and methanol extract was collected. The chloroform, ethyl acetate, methanol and water extracts were taken for the analysis of their antibacterial activity.
4.3.4 TEST MICROORGANISMS USED FOR THE STUDY:
The bacterial strains used for the experiment were collected as pure cultures from the Institute of Nutrition and Food Science (INFS), University of Dhaka. Both Gram positive and Gram-negative organisms were taken for the test and they are listed in the Table-5.1.
TABLE-4.1: LIST OF BACTERIA USED FOR THE TEST.
Gram positive Bacteria Gram negative Bacteria
Staphylococcus aureus Escherichia coli
Vibrio cholerae
Shigella dysenteriae
4.3.5 CULTURE MEDIUM AND THEIR COMPOSITION
The following media is used normally to demonstrate the antimicrobial activity and make subculture of the test organisms.
TABLE-4.2: THE COMPOSITION OF THE AGAR MEDIA USED IN ANTIBACTERIAL ACTIVITY.
Nutrient agar media (gL-1)
Peptone 5
Sodium chloride 5
Beef extract 5
Agar 15
Final pH 7
TABLE-4.3: THE COMPOSITION OF THE MULLER HUNTON MEDIA USED IN ANTIBACTERIAL ACTIVITY.
Muller Hunton media (gL-1)
Peptone 17.5
Beef infusion solid 2.0
Starch 1.5
Agar 17.0
Final pH 7.3±0.2
The media were prepared and sterilized by autoclaving at 121º C for one hour. After sterilization these media were poured in sterile Petri dishes and solidified.
4.3.5.1 PREPARATION OF MEDIUM
To prepare required volume of this medium, calculated amount of each of the constituents was taken in a conical flask and distilled water was added to it make the required volume. The contents were heated in a water bath to make a clear solution. The pH (at 25 oC) was adjusted at 7.2-7.6 using NaOH or HCl. 10 mL and 5 mL of the medium was then transferred in screw cap test tubes to prepare plates and slants respectively. The test tubes were then capped and sterilized by autoclaving at 15 lbs. Pressure/sq. inch at 121 oC for 20 minutes. The slants were used for making fresh culture of bacteria that was in turn used for sensitivity study.
4.3.6 STERILIZATION PROCEDURE
In order to avoid any type of contamination and cross contamination by the test organisms the antimicrobial screening was done in Laminar Hood and all types of precautions were highly maintained. UV light was switched on one hour before working in the Laminar Hood. Petri dishes and other glass wares were sterilized by autoclaving at a temperature of 121oC and a pressure of 15-lbs./sq. inch for 20 minutes. Micropipette tips, cotton, forceps, blank discs etc. were also sterilized.
4.3.7 PREPARATION OF SUBCULTURE
In an aseptic condition under Laminar air cabinet, the test organisms were transferred from the pure cultures to the agar slants with the help of transfer loop to have fresh pure cultures. The inoculated strains were then incubated for 24 hours at 37 0C for their optimum growth. These fresh cultures were used for the sensitivity test.
4.3.8 PREPARATION OF THE TEST PLATES
The test organisms were transferred from the subculture to the test tubes containing about 10 mL of the melted and sterilized agar medium with the help of a sterilized transfer loop in an aseptic area. The test tubes were shaken by rotation to get a uniform suspension of the organisms. The bacterial suspension was immediately transferred to the sterilized petridishes were distribution of the test organisms in the media.
4.3.9 PREPERATION OF THE SAMPLE DISCS
Three types of discs were used for antimicrobial screening.
4.3.9.1 STANDARD DISCS
These were used as positive control to ensure the activity of standard antibiotic against the rest organisms as well as for comparison of the response produced by the known antimicrobial agent with that of the test sample. In this investigation, doxycillin (30μg/disc) standard disc was used as the reference.
4.3.9.2 BLANK DISCS
These were used as negative controls that ensure that the residual solvent (left over the discs even after air-drying) and the filter paper were not active themselves.
4.3.9.3 PREPARATION OF SAMPLE DISCS WITH TEST SAMPLES
Measured amount of each test sample was dissolved in specific volume of solvent to obtain the desired concentrations in an aseptic condition. Sterilized material (BBL, Cocksville, USA) filter discs were taken in a blank petridish under the Laminar hood. Then discs were soaked with solutions of test samples and dried.
4.3.9.3.1 PREPARATION OF SAMPLE DISCS WITH TEST SAMPLES OF POLYGONUM PLEBEJUM
The ethyl acetate extract, methanol extract chloroform extract and water extract were tested for antimicrobial activity against a number of both gram-positive and gram-negative bacteria. The amount of sample per disc was 50 μg/mL
3.3.10 PREPARATION AND APPLICATION OF THE TEST SAMLES
The test samples were weighed accurately and calculated amounts of the solvents were added accordingly using micropipette to the dried samples to get desired concentrations. The test samples were applied to previously sterilized discs using adjustable micropipette under aseptic conditions.
4.3.10.1 DIFFUSION AND INCUBATION
The sample discs, the standard antibiotic discs and the control discs were placed gently on the previously marked zones in the agar plates pre-inoculated with test bacteria. The plates were then kept in a refrigerator at 4 oC for about 24 hours upside down to allow sufficient diffusion of the materials from the discs to the surrounding agar medium. The plates were then inverted and kept in an incubator at 37 oC for hours.
4.3.11 ASSAY OF ANTIMICROBIAL ACTIVITY
4.3.11.1 ANTIBACTERIAL ACTIVITY OF CHLOROFORM, ETHYL ACETATE, METHANOL AND WATER EXTRACT
The chloroform, ethyl acetate and methanol extracts were tested for antibacterial activity by customized Kirby-Bauer disc diffusion. Paper discs were soaked with chloroform (150 µL) extract at concentration (50 mg/mL). Paper discs were also soaked with ethyl acetate (150 µL) and methanol extract separately at concentration (50 mg/mL). 18 hours old bacterial cultures of interest were prepared in Nutrient Broth. Mueller-Hinton agar plates were seeded with the suspensions by sterile cotton buds to yield lawns of the bacteria. The discs were applied on the surface of the plates aseptically using sterile forceps. The discs were pressed gently against the agar by the blunt end of the forceps. The plates were put in incubation in inverted position after 30 minutes. The plates were examined for zone of inhibition after 24 hours incubation at 37ºC.
4.3.12 ANTIBACTERIAL ACTIVITY OF WATER EXTRACT
The antibacterial activity of water extract was assessed by Agar-diffusion method. Solution of water-soluble extract was prepared in sterile distilled water at concentration of 50 mg/mL.
18 hours old bacterial cultures of interest were prepared in nutrient Broth. Mueller-Hinton Agar plates were seeded with suspensions by cotton buds to yield lawns of bacteria. Sterile borer made wells in the agar plates aseptically. Solutions of the extracts in water were dispensed into the wells at 60µL/well. The plates were put in incubation after 30 minutes. The plates were examined for zone of inhibition after 24 hours incubation at 37ºC.The zone diameters were examined under magnifying glass and were measured by a millimeter scale. In every case of positive and negative controls were used. Negative control contained no water extract.
4.4 OBSERVATION AND RESULTS
The dried powder of different herbs was extracted with chloroform, ethyl acetate, methanol and water serially to yield crude organic compounds. The extracts were then dried in a rotavapor at below 40ºC.
Antibacterial activity of different extracts of Khudi Bishkatali (Polygonum plebejum) on different bacteria were studied and some positive results were observed (Table- ). The Table- indicated that the water extracts of Bishkatali (Polygonum plebejum) was active against S. aureus. Other extracts were not active against the bacteria tested.
SUMMARY
Bangladesh is a storehouse of numerous kinds of plants and most of them have medicinal value. Polygonum plebejum is one of the important plants of Polygonaceae family. The natives take this plant as leafy vegetable and used for the remedy of different diseases.
For the present work, the plant was collected from low land area of Mohammadpur. The collected plant was cut into small pieces, dried in an oven at 40oC and powdered mechanically; this powdered sample was extracted with petroleumether and followed by ethylacetate exhaustively. The extracts were concentrated separated to a dry mass. The dry mass (8.683 g) of the ethylacetate extract was subjected to VLC for separation of different compounds.
Phytochemical screening of this plant was carried out from its aqueous extract and taking the powdered specimens using standard procedures. This screening indicated the presence of Alkaloid, Tannin, Saponin, Steroid, Terpenoid, and Cardiac glycoside and quantitative estimation of Alkaloid, Flavonoids, Saponin were carried and their percentage were found to be 0.8, 14.06, 8.5 respectively.
The fractions obtained from ethylacetate extract by VLC using different ratio of elutions were marked as F1, F2, F3, F4, F5, F6, F7, and F8. These fractions were analyzed separately from the fraction F2 and F5 yielded single compounds. Fraction F2 gave single compound (0.892 mg). Its IR was recorded and due to its small amount, it was not further studied.
The fraction F6 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when a solid (4.106 mg) was appeared. It was again dissolved in methanol and its TLC studies showed that it was a single compound with Rf value (0.5) having light yellow crystal.
The fraction F3 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when a solid (6 mg) was appeared. It was again dissolved in methanol and its TLC studies showed that it was mixture compound with light yellow oily substances. Due to lack of time, further study was not possible. Only IR spectrum analysis was carried out.
The fraction F7 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when greenish gummy material was appeared. It was again dissolved in methanol and its TLC studies showed that it was mixture compound with gummy substances. Due to lack of time, further study was not possible. Only IR spectrum analysis was carried out.
Attempt was taken to characterize this Compound (SPPA1) by M.P., IR , 1H-NMR & 13C-NMR spectral data and it was tentatively suggested to be 2,6-dihydroxy-3-acetyl-4-methyl-cyclohex-4-enyl isostearate.
Fatty acids (free fatty acids and bound fatty acids) of the Polygonum plebejum were analyzed. The compositional analysis of petroleumether extract of Polygonum plebejum by GC revealed that the bound fatty acid portion of this plant was rich in unsaturated fatty acids (such as Stearic acid, Oleic acid, Myristic acid, Lauric acid and Lignoceric acid). No free fatty acid was identified, may due to its trace amount.
The percentages of ash and moisture in this plant of Polygonum plebejum were determined by standard method and they were found to be 14.89 % and 14.04 % respectively in dry powder basis.
The anti-microbial tests of petoleumether extract, ethylacetate extract, methanol extract and water extract of Polygonum plebejum were carried out against 4-types of bacteria by disc diffusion method. All extracts except water extract were found to be inactive against these test organisms.
Fatty acids and ash analysis of Polygonum plebejum indicated that this plant may taken a diet as vegetable. Its anti-microbacterial analysis indicated its medicinal importance. It’s further phytochemical analysis studies can lead to isolation of some new compounds, which may have therapeutic value. On the basis of findings, it can be concluded that this plant Polygonum plebejum has importance as a diet as well as its medicinal value.
SUMMARY
Bangladesh is a storehouse of numerous kinds of plants and most of them have medicinal value. Polygonum plebejum is one of the important plants of Polygonaceae family. The natives take this plant as leafy vegetable and used for the remedy of different diseases.
For the present work, the plant was collected from low land area of Mohammadpur. The collected plant was cut into small pieces, dried in an oven at 40oC and powdered mechanically; this powdered sample was extracted with petroleumether and followed by ethylacetate exhaustively. The extracts were concentrated separated to a dry mass. The dry mass (8.683 g) of the ethylacetate extract was subjected to VLC for separation of different compounds.
Phytochemical screening of this plant was carried out from its aqueous extract and taking the powdered specimens using standard procedures. This screening indicated the presence of Alkaloid, Tannin, Saponin, Steroid, Terpenoid, and Cardiac glycoside and quantitative estimation of Alkaloid, Flavonoids, Saponin were carried and their percentage were found to be 0.8, 14.06, 8.5 respectively.
The fractions obtained from ethylacetate extract by VLC using different ratio of elutions were marked as F1, F2, F3, F4, F5, F6, F7, and F8. These fractions were analyzed separately from the fraction F2 and F5 yielded single compounds. Fraction F2 gave single compound (0.892 mg). Its IR was recorded and due to its small amount, it was not further studied.
The fraction F6 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when a solid (4.106 mg) was appeared. It was again dissolved in methanol and its TLC studies showed that it was a single compound with Rf value (0.5) having light yellow crystal.
The fraction F3 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when a solid (6 mg) was appeared. It was again dissolved in methanol and its TLC studies showed that it was mixture compound with light yellow oily substances. Due to lack of time, further study was not possible. Only IR spectrum analysis was carried out.
The fraction F7 was concentrated to a small volume in vacuum and left undisturbed at room temperature for several days when greenish gummy material was appeared. It was again dissolved in methanol and its TLC studies showed that it was mixture compound with gummy substances. Due to lack of time, further study was not possible. Only IR spectrum analysis was carried out.
Fatty acids (free fatty acids and bound fatty acids) of the Polygonum plebejum were analyzed. The compositional analysis of petroleumether extract of Polygonum plebejum by GC revealed that the bound fatty acid portion of this plant was rich in unsaturated fatty acids (such as Stearic acid, Oleic acid, Myristic acid, Lauric acid and Lignoceric acid). No free fatty acid was identified, may due to its trace amount.
The percentages of ash and moisture in this plant of Polygonum plebejum were determined by standard method and they were found to be 14.89 % and 14.04 % respectively in dry powder basis.
The anti-microbial tests of petoleumether extract, ethylacetate extract, methanol extract and water extract of Polygonum plebejum were carried out against 4-types of bacteria by disc diffusion method. All extracts except water extract were found to be inactive against these test organisms.
Fatty acids and ash analysis of Polygonum plebejum indicated that this plant may taken a diet as vegetable. Its anti-microbacterial analysis indicated its medicinal importance. It’s further phytochemical analysis studies can lead to isolation of some new compounds, which may have therapeutic value. On the basis of findings, it can be concluded that this plant Polygonum plebejum has importance as a diet as well as its medicinal value.