Main purpose of this thesis is to analysis of Escherichia coli and Shigella species in the Freshwater Environment of Bangladesh by PCR-DGGE. Other objectives are Recovery of the environmentally stressed, injured and starved Shigella spp. and E. coli from freshwater environment. Here also Statistical analysis to correlate the occurrence of different phyla, genes and bacterial genera with one another. Finally identification of the community of E. coli and Shigella spp. by assessing the sequencing results of the DGGE bands, results of PCR assay for specific virulent genes and the outputs of statistical analysis.
Introduction
Only about 6 years ago, the knowledge of bacteria that naturally occur in freshwater ecosystems was restricted to organisms that could be grown in culture. However, it was known that about 98% bacteria in the environment could not be cultured. Recently, several studies have examined bacterial community composition in the epilimnion of freshwater lakes and reservoirs. The major bacterial phyla represented in most or all freshwater sites are the Proteobacteria (alpha, beta and gamma subdivisions), the Cytophaga–Flavobacterium–Bacteroides (CFB) group, the Actinobacteria, the Cyanobacteria and the Verrucomicrobia (Zwart et al., 2002).
About 400 children below the age 5 die per hour in the developing world from waterborne diarrheal diseases (Gadgil, 1998). Pathogenic E. coli and Shigella species are two most important water-related pathogens (Egli et al., 2002). World Health Organization (WHO, 2001) has categorized the diseases caused by Shigella and Enterohemorrhagic E. coli (EHEC) as priority waterborne disease.
Shigellosis is endemic throughout the world, but 99% of the 150 million annual cases and almost all of the million deaths occur in the developing world (Kotloff et al., 1999). Pathogenic E. coli strains are a major cause of infant diarrhea in developing countries, particularly in the 0- to 6-month age group.
There is evidence that Escherichia coli will survive indefinitely in tropical waters and may even multiply (Carillo et al., 1985). However, identification of Shigella in environmental samples is limited mainly by the lack of a suitable enrichment technique (Faruque et al., 2002). Although contaminated water is a major causative agent for shigellosis, until now there is little or no credible report on isolation of Shigella from aquatic environment and its reproducibility. Probably, the shigellae are fragile or might remain in a state of dormancy or might be starved, injured or stressed. In water, there may be lack of appropriate nutrients or growth factors inculding the physiological factors that may not be favourable for cell resuscitation or repair and hence recovery. The recovery and detection of these Shigella spp. becomes difficult employing conventional cultural techniques followed by molecular analysis.
This has implied the importance of the community structure analysis of E. coli and Shigella species. This type of analysis helps to determine the interactive members in the habitat which are always present with these bacteria. Identification of such stable community may be helpful to detect the presence of such bacteria that resist cultivation, which is an essential prerequisite for characterization in the laboratory. Community structure analysis of E. coli and Shigella species may be also helpful to observe the recovery capacity of pre-enrichment and enrichment techniques used to recover specially these bacteria.
The nucleic acid sequence comparisons are fundamental and straightforward way to classify and relate organisms now a day. A number of methods have been developed that exploit the sequence divergence among taxa to examine microbial community structure. These culture-independent methods for microbial community analysis most often utilize polymerase chain reactions (PCR) to amplify phylogenetic markers such as highly variable V3 region of the bacterial 16S rDNA gene in DNA extracted from the microbial community (Wilson et al., 1997).
Denaturing gradient gel electrophoresis (DGGE) is one of such methods that are commonly used. With DGGE many samples can be analyzed and it has the ability to tailor the analysis to examine particular organisms or taxa of interest through the use of universal or group-specific primers. PCR can also be used to identify the presence of pathogens by detecting specific virulent genes.
Literature Review
Microbial communities
The community is the highest biological unit in an ecological hierarchy made up of individuals and populations (Figure 1.1). Populations within a community interact with each other in an integrated manner. They do so at a physical location called a habitat. Some microorganisms are autochthonous or indigenous within a given habitat, which are capable of survival, growth, and metabolic activity in that habitat. In contrast, allochthonous microorganisms are transient members of their habitat (Atlas and Bartha, 1998).
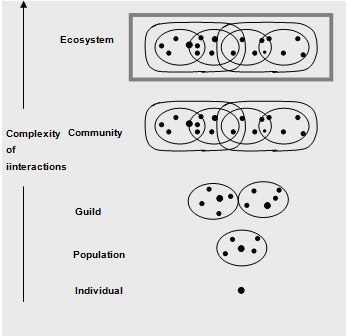
The habitat is one component of a broader concept known as the ecological niche, which includes not only where an organism lives but also what it does there; the niche is the functional role of an organism within an ecosystem. The niches within a community are filled by the indigenous populations of that community. Microbial populations exhibit various adaptations for success in diverse communities. These adaptations as well as population interactions contribute to the stability of communities. Stable microbial communities tend to have high diversities. Populations within a community that use the same resources (the guild structure of the community) often exhibit intense competition. In some cases the first organisms to arrive and colonize an area having a selective advantage and can retain a niche in the community against competitors. In other cases there is a succession of populations, with better adapted populations displacing those originally occupying a niche.
The species assemblage that successfully inhabits a delineated volume of resources is called a unit community. Populations in a unit community tend to interact with each other and not with populations in other unit communities (Swift, 1984).
Diversity and stability of microbial communities
Traditionally, the unit of diversity is the species, and a variety of definitions for the concept are used for these organisms. First, the “phylophenetic” definition circumscribes the species as a “monophyletic and genomically coherent cluster of individual organisms that show a high degree of overall similarity in many independent characteristics, and is diagnosable by a discriminative phenotypic property”. Second, a species can be defined as an assemblage of strains sharing 70% or more DNA homology. Third, in an ecological definition the species and niche concept are linked, and thus a species consists of the organisms occupying the same niche. Thus, diversity can be defined as the number of prokaryotic species and their relative abundance in a community, or as the amount and distribution of information in a community (Torsvik et al., 2002).
Dynamics and Control of Prokaryotic Diversity
Trophic interactions: Among prokaryotes, competitors can coexist if some mechanism of selective loss is operating to prevent the most successful competitors from sequestering all the resources. For example, parasitism by host-specific viruses will allow coexistence of different bacterial taxa within the bacterial community.
Evolutionary perspective: The ecological factors and the intrinsic evolutionary mechanisms working at molecular and population levels interact to control prokaryote diversity. Microbial diversity in soil ecosystems exceeds, by far, that of eukaryotic organisms. One gram of soil may harbor up to 10 billion microorganisms of possibly thousands of different species (Torsvik and Øvreås, 2002). One reason for the high genomic diversity observed in prokaryotic communities in soil and sediments is the large populations of organisms and the capacity to accumulate large numbers of mutations.
Temporal heterogeneity: Most terrestrial communities intermittently suffer disturbances, such as starvation, desiccation, freezing/thawing, or human activity. Altered environmental conditions and resource availability create opportunities for new species to become established, and disturbances will ensure that communities include a mixture of different stages of succession.
Spatial heterogeneity: The structural complexity of habitats is important for population-level diversification because it allows resources to be partitioned and creates new niches, thereby enhancing prokaryote specialization and division into distinct ecological species (Torsvik et al., 2002).
Freshwater microbial communities
Freshwater habitats
Freshwater habitats are classified based on their chemical and physical properties. Those with standing water, such as lakes and ponds, are called lentic habitats; those with running water are lotic habitats (Wetzel 1975).
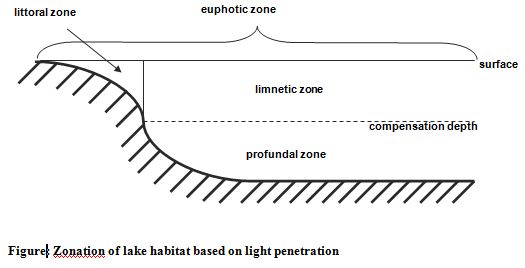
Lakes: Lakes are divided into three zones based on the penetration of light (Figure 1.2). The combined littoral and limnetic zones are known as the euphotic zone; here photosynthetic activity can occur. The profundal zone is the area of deeper water beyond the depth of effective light penetration; it does not exist in shallow ponds. In deep lakes the profundal zone extends from the light compensation level to the bottom (Atlas and Bartha, 1998).
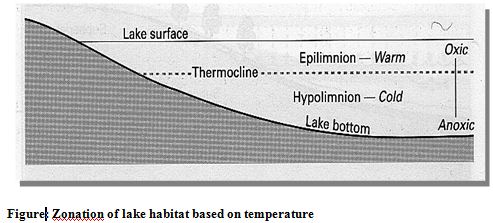
On the basis of temperature lakes can be divided into three zones (Figure). The epilimnion is the upper warm zone of water which is separated from the cold deep hypolimnion by the thermocline. The thermocline is a zone characterized by rapid decrease in temperature, across which there is little mixing of water.
An ecologically useful classification of lake habitats as oligotrophic or eutrophic is based on productivity and nutrient concentrations. Oligotrophic lakes have low concentrations of nutrients. Typically, they are deep, have a large hypolimnion than epilimnion, and have relatively low primary productivity. In contrast, eutrophic lakes have high nutrient concentrations, are usually shallower and warmer than oligotrophic lakes, and have higher rates of primary production (Atlas and Bartha, 1998)
Composition and activity of freshwater microbial communities
Bacterioplankton communities are integrally involved in the biogeochemical processes underpinning freshwater ecosystems (Newton et al., 2006). The principal ecological functions of microorganisms in freshwater environments can be summarized as follows: (1) they decompose dead organic matter; liberating mineral nutrients for primary production (2) they assimilate and reintroduce into the food web dissolved organic matter (3) they perform mineral cycling activities (4) they contribute to primary production (5) they serve as a food source for grazers (Atlas and Bartha, 1998).
Community composition determined by cultural techniques
The microbial populations of lakes have been much more extensively studied than those of rivers. Members of the genera Achromobacter, Flavobacterium, Brevibacterium, Micrococcus, Bacillus, Pseudomonas, Nocardia, Streptomyces, Micromonospora, Cytophaga, Spirillum, and Vibrio are reported as occurring widely in lake water. Stalked bacteria, such as Caulobacter, Hyphomicrobium, and other genera, are associated many with submerged surfaces (Rheinheimer, 1991).
Autotrophic bacteria are autochthonous member of the microbiota of lakes and play an important role in nutrient cycling. Photoautotrophic bacteria normally found in lakes include cyanobacteria and in anoxic zones the purple and green anaerobic photosynthetic bacteria. The cyanobacteria Microcystis, Anabaena, and Aphanizomenon can be dominant plankton in freshwater habitats. Chemolithotrophic bacteria have important roles in nitrogen, sulfur and iron cycling within lakes; members of the genera Nitrosomonas, Nitrobacter, and Thiobacillus are essential members of freshwater microbial communities.
In addition to autochthonous microbial populations, many allochthonous terrestrial microorganisms are carried by erosion and runoff from soils into freshwater aquatic ecosystems. Allochthonous microorganisms also enter when leaves from adjacent plants fall into these water bodies and when municipal sewage enters freshwater environments together with high amounts of organic matter. Heterotrophic microbial populations in areas that receive high amounts of organic matter are generally elevated, but as the amounts of imported organic matter decrease, populations of heterotrophic microorganisms also decline (Atlas and Bartha, 1998).
Community Composition determined by molecular techniques
Only about 6 years ago, our knowledge of bacteria that naturally occur in freshwater ecosystems was restricted to organisms that could be grown in culture. However, it was known that most bacteria in the environment could not be cultivated. In fact, on the basis of cultural techniques Rheinheimer (1991) concluded that bacteria found in groundwater, spring water and streams also occur in soils, and that there was no clear separation between soil bacteria and aquatic bacteria. These observations call into question the existence of a unique freshwater bacterial flora.
Recently, several studies have examined bacterial community composition in the epilimnion of freshwater lakes and reservoirs. As a result of these studies, a core group of bacterial phylotypes common to freshwater has emerged. The major bacterial divisions represented most or all freshwater sites are the Proteobacteria (alpha, beta and gamma subdivisions), the Cytophaga–Flavobacterium–Bacteroides (CFB) group, the Actinobacteria, the Cyanobacteria and the Verrucomicrobia (Zwart et al., 2002).
Another study identified representatives of six bacterial phyla e.g. Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, TM7 and Verrucomicrobia, including members of the classes Alpha-, Beta-, Delta and Gammaproteobacteria, as well as phylotypes with unknown affiliation in a freshwater site (Newton et al., 2006).
Sources of variation in bacterial community composition
There is tremendous variability in the composition of communities of bacterioplankton among lakes. A number of important parameters make lakes more or less suitable habitats for microorganisms. Examination of these factors that regulating not only the microbial community dynamics, but the dynamics of individual members of that community will lead to an increased understanding of potential freshwater microbial-mediated processes linked to specific organisms (Newton et al., 2006).
Geographic sources of variation: Regionalization has important influence on bacterial community composition (Lindström and Leskinen, 2002). There are several factors that could account for these regionalized effects including geology, climate and vegetation, land use and land cover, and anthropogenic impacts on lakes.
Environmental sources of variation: Two principal ecological forces, one related to lake primary productivity and one related to dissolved organic carbon (DOC) (Yannarell and Triplett, 2004) were significantly related to variation in bacterial community composition. Additionally, concentrations of inorganic nutrients, pH, salt concentrations, water clarity and concentrations of oxygen also have important impact on bacterial community composition.
Temporal variation: Lake bacterial communities show considerable variation in time and exhibit seasonal patterns. The temporal variation of bacterial community composition is controlled by a tight set of constraints. These constraints may include lower water temperatures; the presence or absence of large populations of grazers (i.e., the clear-water phase); the activity of primary producers; and the concentrations of limiting nutrients (Yannarell and Triplett, 2005).
Important waterborne pathogens: Escherichia coli and Shigella spp.
Diarrheal diseases are one of the major waterborne diseases in the developing countries. About 400 children below the age 5 die per hour in the developing world from waterborne diarrheal diseases. Waterborne infectious diseases are transmitted primarily through contamination of the water sources with excreta of humans and animals who are either active cases or carriers of the disease. Use of such water for drinking or cooking, contact with it during washing or bathing, or even inhalation of its fine droplets as aerosols, may then result in infection (Gadgil, 1998).
Pathogenic E. coli and Shigella species are two most important waterborne pathogens (Egli et al., 2002). World Health Organization (WHO, 2001) has categorized the diseases caused by Shigella and Enterohemorrhagic E. coli (EHEC) as priority waterborne disease.
Shigella species
Bacillary dysentery is an acute inflammatory bowel disease caused by an infectious agent, the enteroinvasive genus Shigella, hence the name shigellosis. Shigellosis is endemic throughout the world, but 99% of the 150 million annual cases and almost all of the million deaths occur in the developing world, particularly in areas where personal and general hygiene are inadequate and poor. Shigellosis is a disease of impoverished people which in about 70% of the cases affects children between the ages of 1 and 5 years (Kotloff et al., 1999).
Shigellosis occurs as an endemic disease in Bangladesh, and at least three large epidemics caused by Shigella dysenteriae type 1 have occurred between 1972 and 1994, causing high morbidity and mortality, particularly in children (Chen et al., 1980, Ronsmans et al., 1988 and Katz, 1986). There are four species of Shigella: S. dysenteriae, S. flexneri, S. sonnei and S. boydii (Hale, 1991).
Pathogenesis of bacillary dysentery
Shigella is a highly contagious microorganism since as few as 10-100 bacteria can cause the disease in adult volunteers. After oral contamination, bacteria pass through the stomach and the small intestine before reaching the colon where they invade the mucosa, initiating the acute destructive recto-colitis that causes the dysenteric symptoms: fever, intestinal cramps and emission of mucopurulent and bloody stools. The basis for organ specificity of shigellosis to the rectal and colonic mucosae is not understood. Shigella may express a colon-specific adhesive system, or the colonic and rectal mucosae may be more susceptible to developing acute inflammation in the presence of invasive shigellae. The disease remains essentially limited to the intestinal mucosa and septicemic dissemination is a rare event, except in malnourished children. The molecular and cellular effectors of innate immunity that eradicate the bacteria during the phase of primary infection and prevent systemic dissemination at the price of intestinal tissue destruction have yet to be fully identified and their mode of action characterized. Shigellosis can be seen as a loss of balance in the host mechanisms that regulate inflammation in the presence of an invading microorganism. A series of discoveries have recently allowed progress in the understanding of the molecular mechanisms by which Shigella disrupts, invades and destroys the intestinal barrier (Hale, 1998).
Determinants of pathogenesis expressed by Shigella
Plasmid genes: All virulent strains of Shigella carry a 220-kb plasmid which encodes the `invasive phenotype’ of this species. The coding sequences are scattered on the entire plasmid with one block of 30 kb showing a particularly dense pattern of genes, the ipa/mxi-spa locus that can be considered the central S. flexneri pathogenicity island (PAI). This PAI (Figure 1.4) is necessary and sufficient to cause entry of Shigella into epithelial cells and macrophage apoptotic death. Based on available data, one can consider that this PAI primarily encodes a type III secreton, in other words a flagella-like structure able to deliver Shigella proteins straight from the bacterial cytoplasm into the cytoplasmic membrane of epithelial cells, or into their cytoplasm.
At least five proteins encoded by the mxi operon assemble to form the secretory apparatus. There are about 20 candidate target proteins secreted through this secreton, upon contact of the bacterium with the epithelial cell surface. Five of them, IpaA-D and IpgD, are encoded by the 30-kb PAI. Others are encoded by genes scattered on the virulence plasmid (Sansonetti, 2001).
There are two categories of proteins secreted through the type III secreton:
IpaB, IpaC and IpaD are essential to the initial events of secretion. IpaB and IpaD form a complex which controls the flux of proteins through the type III secreton. IpaA and IpgD, like IpaB, C and D, are constitutively expressed at 37oC, regardless of the activity of the type III secreton. Deletion of their respective genes does not eliminate the entry capacity of the mutants, but induces significant attenuation, indicating that these genes have an effect on the maturation of the entry focus.
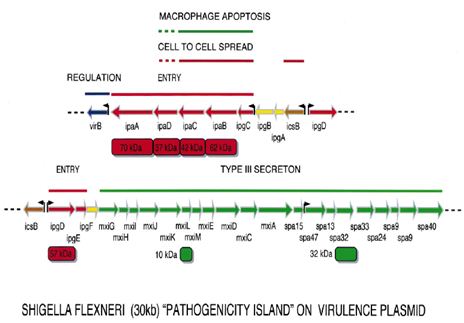
Figure: Map of the plasmid-located Shigella `pathogenicity island’ of 30 kb that is required for entry into epithelial cells and killing of macrophages
The second category of proteins encompasses two subgroups. These proteins, of still unidentified function, correspond to putative additional targets for the type III secreton, such as members of the IpaH family, SopB and VirA. Their genes are transcriptionally induced upon activation of the secreton. In any event, the type III secreton and its target proteins can now be regarded as the major weapon that Shigella uses to enter into epithelial cells and also to alter the function of others such as phagocytes.
Beside the type III secreton and its cognate target proteins, other important proteins are encoded by the virulence plasmid. Among these, SepA is a secreted serine protease whose function has not yet been established. The most important, however, is IcsA (VirG), which achieves the actin-based motility of Shigella and permits its passage from one cell to another. IcsA (VirG) is a 120-kDa surface protein which localizes at one pole of the bacterial body and is able to cause actin-dependent motility. IcsA mutants are severely impaired in pathogenicity, including in monkeys and even in human volunteers as recently tested (Sansonetti, 2001).
Chromosomal genes associated with virulence: Shigella is an interesting paradigm of co-evolution and mutual adaptation between plasmid and chromosomal virulence genes. Beside the core of virulence plasmid genes that dictate the direct interaction of bacteria with the various cell populations that make up the epithelial barrier, chromosomal genes also participate in the pathogenic process.
These chromosomal genes can be classified into two categories: (i) genes that regulate the expression of the virulence genes on the plasmid. This category is exemplified by virR, a gene encoding a histone-like molecule which controls the temperature-dependent expression of Ipa and Mxi-Spa proteins; (ii) genes that are important for bacterial survival in the intestinal tract and in infected tissues such as those encoding the LPS and siderophores, several of them being located on increasingly identified PAIs. In addition, in S. dysenteriae 1, shiga toxin (stx) is encoded by a chromosomal locus (Sansonetti, 2001).
Escherichia coli
Most strains of E. coli are harmless commensal members of intestinal flora of mammals and, to undetermined extent, birds in which some strains adhere to the intestinal mucosa while others are only temporary residents in the lumen of colon. Secondary habitats are soil, sediment and water (Savageau, 1983), where its density is proportional to the amount of fecal contamination. E. coli is an indicator organism used worldwide, thus the presence of E. coli in water is regarded as a warning signal: the water is subjected to potentially dangerous pollution. Three general clinical syndromes result from infection with inherently pathogenic E. coli strains: (i) urinary tract infection, (ii) sepsis/meningitis, and (iii) enteric/ diarrheal disease (Nataro and Kaper, 1998).
Common themes in E. coli virulence:
On the basis of virulence factors currently, at least six virotypes of E. coli have been identified: enterotoxigenic E. coli (ETEC), enteropathogenic E. coli (EPEC), enterohemorrhagic E. coli (EHEC), enteroaggregative E. coli (EAEC), enteroinvasive E. coli (EIEC), diffusely adherent E. coli (DAEC) (Salyers and Whitt, 2002). The characteristics that form the basis for the ‘virotyping’ system include patterns of bacterial attachment to host cells, effects of attachment on host cells, production of toxins and invasiveness. Like most mucosal pathogens, E. coli can be said to follow a requisite strategy of infection: (i) colonization of a mucosal site, (ii) evasion of host defenses, (iii) multiplication, and (iv)host damage(Nataro and Kaper, 1998). However, the interaction of the organisms with the intestinal mucosa is specific for each category. Schematized paradigms are illustrated in Figure.
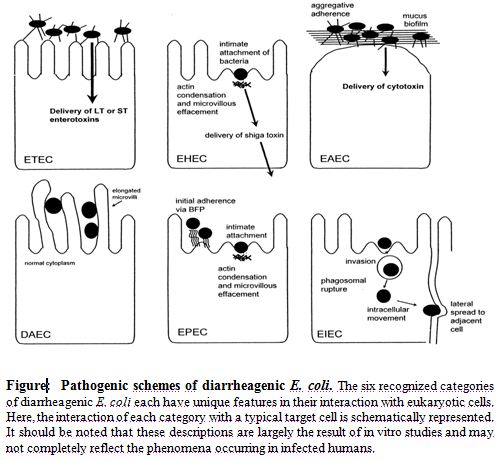
The E. coli virotypes
Enterotoxigenic E. coli (ETEC) is catagorised as containing the E. coli strains that show at least one member of two defined groups of enterotoxins: ST and LT (Johnson et al., 1990). ETEC strains are associated with two major clinical syndromes: weanling diarrhea among children in the developing world, and travelers’ diarrhea. Epidemiologic investigations have implicated contaminated food and water as the most common vehicles for ETEC infection (Nataro and Kaper, 1998).
In contrast to the limited importance of enteropathogenic E. coli (EPEC) in developed countries, EPEC is a major cause of infant diarrhea in developing countries. Particularly in the 0- to 6-month age group, EPEC strains are often the most frequently isolated bacterial diarrheal pathogens. In 1992, Donnenberg and Kaper (Donnenberg and Kaper, 1992) proposed a threestage model of EPEC pathogenesis consisting of (i) localized adherence, (ii) signal transduction, and (iii) intimate adherence.
Enterohemorrhagic E. coli (EHEC) strains have only recently been recognized as a cause of serious disease. In the EHEC group of strains, there is one predominant serogroup and serotype (0157:H7) (Salyers and Whitt, 2002). Although EHEC strains cause dysentery similar to that caused by Shigella spp., they probably do not invade mucosal cells as readily as Shigella strains. A toxin produced by EHEC strains is virtually identical to shiga toxin, the major virulence factor, and a defining characteristic of EHEC, is Stx (Nataro and Kaper, 1998).
Enteroaggregative E. coli (EAggEC) strains, the newest virotype, cause a persistent form of diarrhea in children. EAggEC strains produce an ST-like toxin and a hemolysin-like toxin. EAEC has been implicated as a cause of sporadic diarrhea in Mexico, Chile, Bangladesh, and Iran (Brenner et al., 1973). Enteroinvasive E. coli (EIEC) strains are biochemically, genetically, and pathogenetically closely related to Shigella spp. EIEC strains are generally lysine decarboxylase negative, nonmotile, and lactose negative (Brenner et al., 1973). Endemic sporadic disease occurs in some areas, generally where Shigella spp. is also prevalent, but the epidemiologic features may be different from those of Shigella spp. (Nataro and Kaper, 1998).
Little is known about the pathogenetic features of diffusely adherent E. coli (DAEC) induced diarrhea. Bilge et al. (1989) have described the cloning and characterization of a surface fimbria in this strain, which mediates the DA phenotype. The genes encoding the fimbria can be found on either the bacterial chromosome or a plasmid. The association of DAEC with diarrhea is found only in children older than infants (Nataro and Kaper, 1998).
Community analysis by 16S ribosomal RNA genes
Nucleic acid sequencing technology is bringing a much-needed phylogenetic perspective into microbiology. There is no more fundamental and straightforward way to classify and relate organisms than by appropriate nucleic acid sequence comparisons. The simple morphology of most microbes provides few clues for their identification; physiological traits are often ambiguous. The microbial ecologist is particularly impeded by these constraints, since only 0.001 to 15% of the total cell counts in environmental samples can be cultivated (Amann et al., 1995).
The use of macromolecular comparisons to infer phylogenetic relationships is now well established. Comparisons may be based either on experimental measurements of “molecular similarity” (e.g. antibody cross reactivity, DNA-DNA hybridization, and ribosomal RNA-DNA hybridization) or on mathematical analyses of molecular sequence data. The former methods require the pairwise experimental comparison of most, or preferably all, organisms considered. In contrast, sequence data are readily accumulated, creating a “data base” that can be referred to for phylogenetic analysis of new sequence data as they become available (Olsen et al., 1986).
The rRNA molecules have long been recognized for their utility as molecular chronometers (Kent and Triplett, 2002). Characterization of unknown organisms by rRNA sequences requires a reference collection of sequences from known organisms. For the analysis of natural microbial populations, in which unknown diversity must be anticipated, there are several reasons to focus on the rRNAs as mentioned below
- The rRNAs, as key elements of the protein-synthesizing machinery, are functionally and evolutionarily homologous in all organisms.
- The rRNAs are ancient molecules and are extremely conserved in overall structure. Thus, the homologous rRNAs are readily identifiable, by their sizes.
- Nucleotide sequences are also conserved. Some sequence stretches are invariant across the primary kingdoms, while others vary. The conserved sequences and secondary structure elements allow the alignment of variable sequences so that only homologous nucleotides are employed in any phylogenetic analysis. The highly conserved regions also provide convenient hybridization targets for cloning the rRNA genes and for primer directed sequencing techniques.
- The rRNAs constitute a significant component of the cellular mass, and they are readily recovered from all types of organisms for accumulation of a data base of reference sequences.
- The rRNAs provide sufficient sequence information to permit statistically significant comparisons.
- The rRNA genes seem to lack artifacts of lateral transfer between contemporaneous organisms. Thus, relationships between rRNAs reflect evolutionary relationships of the organisms (Olsen et al., 1986).
There are three rRNAs in bacteria, 5S (~120 nucleotides), 16S (~1600 nucleotides), and 23S (~3000 nucleotides). The 16S rRNA is appropriate size for broad phylogenetic analysis, but it was too large for complete sequence determinations until the development of DNA cloning and sequencing protocols. Instead, the 16S rRNA was subjected to partial sequence analysis, so-called “oligonucleotide cataloging,” which characterizes ~25% of the sequence as unique RNase T1 oligonucleotides (Olsen et al., 1986).
Culture-independent methods of assessing microbial diversity
PCR-based methods
A number of methods have been developed that exploit the sequence divergence among taxa to examine microbial community structure. These culture-independent methods for microbial community analysis most often utilize polymerase chain reactions (PCR) to amplify phylogenetic markers in DNA extracted from the microbial community.
Community analyses based on PCR have a number of steps that may introduce biases, starting with DNA extraction. Bacterial cell structure varies among taxonomic groups, with some bacteria being more easily disrupted than others. In addition, environmental factors require special consideration for both sample collection and DNA extraction. PCR may be inhibited by the presence environmental compounds such as soil (Wilson et al., 1997).
Despite these problems, PCR-based community analysis methods are commonly used because of the ease with which many samples can be analyzed and the ability to tailor the analysis to examine particular organisms or taxa of interest through the use of universal or group-specific primers. A number of community “fingerprint” methods are commonly used to assess differences in community composition between samples or treatments or to assess changes in microbial communities over time. Such techniques as ribosomal intergenic spacer analysis (RISA), denaturing gradient gel electrophoresis (DGGE), temperature gradient gel electrophoresis (TGGE), single-strand-conformation polymorphism (SSCP), ITS-restriction fragment length polymorphism (ITS-RFLP), random amplified polymorphic DNA (RAPD), or amplified ribosomal DNA restriction analysis (ARDRA) yield complex community profiles that do not directly offer phylogenetic information but do allow analysis and comparisons of community composition. Differences in electrophoretic profiles between samples reflect differences in community composition and abundance of individual microbial populations in a community. Although the fingerprint obtained from an environmental sample cannot reveal the taxonomic composition of a microbial community, phylogenetic information about particular community members may be obtained by isolation and sequence analysis of bands of interest.
While correlations between the distribution of PCR-amplified phylogenetic markers and species distribution have limitations owing to the presence of multiple rRNA operons in bacteria and PCR and cloning biases, molecular methods for community analysis can reveal the presence of microorganisms that remain intractable to traditional cultivation techniques (Kent and Triplett, 2002).
Denaturing Gradient Gel Electrophoresis (DGGE)
Originally DGGE was introduced into microbial ecology to determine the genetic diversity of complex mixtures of bacterial populations. DGGE of PCR-amplified DNA fragments can be used to: (i) study community complexity (ii) monitor population shifts (iii) analyze enrichment cultures and the isolation of bacteria (iv) detect sequence heterogeneities of 16S rRNA genes in single genomes (v) compare DNA extraction methods (vi) screen clone libraries (vii) determine PCR and cloning biases.
In DGGE DNA fragments of the same length but with different base-pair sequences can be separated. Separation in DGGE is based on the decreased electrophoretic mobility of a partially melted DNA molecule in polyacrylamide gels containing a linearly increasing gradient of DNA denaturants (a mixture of urea and formamide). Melting of the DNA fragments proceeds in discrete so-called melting domains: stretches of base pairs with an identical melting temperature. Once a melting domain reaches its melting temperature at a particular position in the denaturant gradient gel, a transition of helical to partially melted molecules occurs, and migration of the molecule will practically halt. Sequence variation within such domains causes their melting temperature to differ. Sequence variants of particular fragments will therefore stop migrating at different positions in the denaturing gradient and hence can be separated by DGGE.
Using this approach, 50% of the sequence variants can be detected in DNA fragments up to 500 bp. This percentage can be increased to nearly 100% by the attachment of a GC-rich sequence to the DNA fragment, which will then act as a high temperature melting domain. Attachment of the GC-clamp can be done by cloning or, if the polymerase chain reaction (PCR) is used to create DNA fragments, by the addition of a 40 bp or 50 bp GC-rich sequence to the 5′ end of one of the PCR primers (Muyzer et al., 2004).
Objectives of the study
General objective
Pathogenic E. coli and Shigella spp. are two most important water-related pathogens (Egli et al., 2002). There is evidence that Escherichia coli will survive indefinitely in tropical waters and may even multiply (Carillo et al., 1985) whereas data relating to Shigella isolation and survival is almost nil. The objectives of this investigation encompass the identification of the community structure of E. coli and Shigella species which may be helpful to detect the presence of such bacteria that resist cultivation particularly Shigella spp. from the aquatic environment.
Specific objectives
- Recovery of the environmentally stressed, injured and starved Shigella spp. and E. coli from freshwater environment. Extraction of total community DNA from raw water samples and also from enriched samples.
- PCR amplification of the highly variable V3 region of the bacterial 16S rRNA gene.
- DGGE analysis of the PCR products and sequence analysis of DGGE fragments to identify the bands.
- PCR assay for the amplification of lt1, st1, bfp, stx1, ipaH, ipaBCD, virA and ial genes specific for Shigella spp. and E. coli.
- Statistical analysis to correlate the occurrence of different phyla, genes and bacterial genera with one another.
- Identification of the community of E. coli and Shigella spp. by assessing the sequencing results of the DGGE bands, results of PCR assay for specific virulent genes and the outputs of statistical analysis.
Results
Microbial populations exhibit various adaptations for survival and activity in diverse communities. These adaptations as well as population interactions contribute to the stability of communities. Diarrheal diseases are one of the major waterborne diseases in the developing countries. Pathogenic E. coli and Shigella spp. are two most important waterborne pathogens (Egli et al., 2002). There is evidence that Escherichia coli will survive indefinitely in tropical waters and may even multiply (Carillo et al., 1985). However, identification of Shigella in environmental samples is limited mainly by the lack of a suitable enrichment technique (Faruque et al., 2002). Detection of a stable community of E. coli and Shigella spp. may be helpful to detect the presence of such bacteria that resist recovery by cultural means e.g. Shigella spp. In the present study PCR-DGGE approach combined with statistical analysis was used to identify the community of E. coli and Shigella spp. in the freshwater environment in Bangladesh.
Analysis of DGGE profiles
To profile community complexity PCR amplified 16S rDNA fragments were separated by DGGE. From the DGGE profiles (Figures 3.1 and 3.2), it is clear that there was a high variability between the bacterial assemblages although some bands are common to several water bodies (e.g. band 4, Figure 3.1)
The major DGGE bands were excised, sequenced and matched with the GenBank database (http://www.ncbi.nih.gov/BLAST). DGGE has some limitations (Muyzer and Smalla, 1998) such as the possible comigration of bands with different sequences. Thus, in order to confirm that matching bands corresponded to identical phylotypes, more than one band with equal position in the gel was excised and sequenced. The same nucleotide sequence was obtained for each band position (e.g. band 4, Figure 3.1). The alignments of the 16S rDNA sequences representing each of the identified genera are given in Appendix-IV.
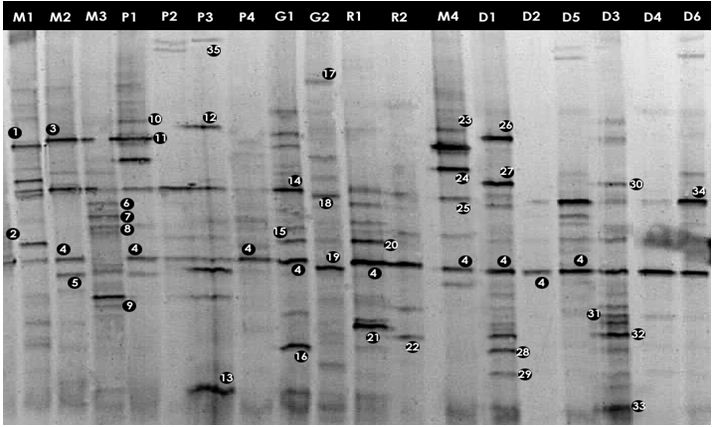
Figure: DGGE gel containing the bacterial 16S rRNA gene fragments obtained after enzymatic amplification of DNA extracted from freshwater samples to assess the bacterial community. The code above each lane refers to each water sample (Table).
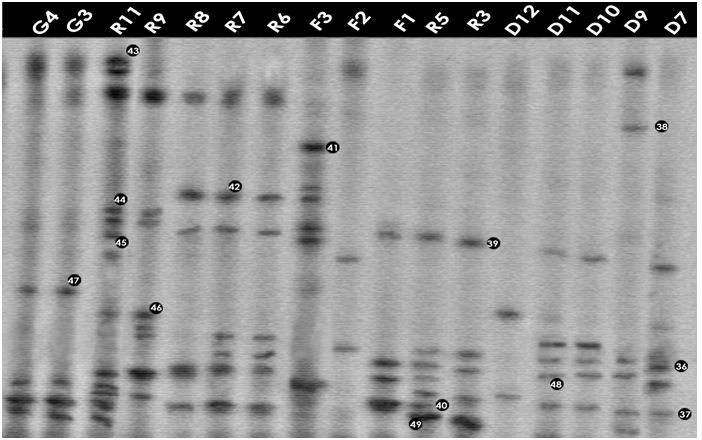
Figure: DGGE gel containing the bacterial 16S rRNA gene fragments obtained after enzymatic amplification of DNA extracted from freshwater samples to assess the bacterial community. The code above each lane refers to each water sample (Table).
Community composition of different lakes
Community composition of Mirpur lake
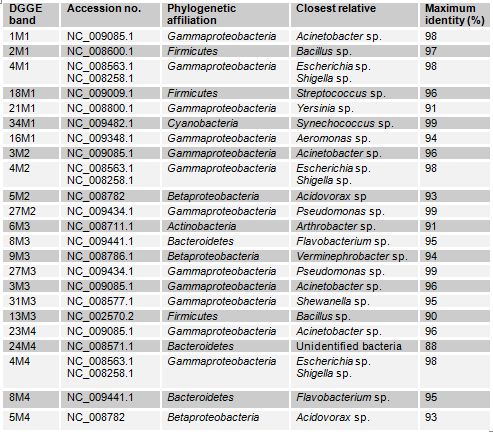
The DGGE profiles (Figure 3.1) of the four samples collected from different sites of Mirpur lake shows the relative composition of the bacterial communities were different. The phylogenetic affiliation (Table 3.1) of each sequenced DGGE band shows that they belong to members of five bacterial phyla: Gammaproteobacteria, Betaproteobacteria, Bacteroidetes, Actinobacteria, Firmicutes and Cyanobacteria. The Gammaproteobacteria was the dominant phylum in this site present in all the four samples more than once. In these samples prominent bands according to band intensity in the DGGE gel (Figure 3.1) were most closely related to known organisms in the database included Acinetobacter sp. (band 1M1), Bacillus sp. (2M1), the Escherichia-Shigella group (4M2), Acidovorax sp. (5M4), Streptococcus sp. (18M1), Synechococcus sp. (34M1), Arthrobacter sp. (6M3), Flavobacterium sp. (8M3) and Verminephrobacter sp. (9M3). One prominent band (24M4) was most closely related to unidentified bacteria at an identity level of 88%. Percent identities ranged between good (99%) and poor (88%).
Community composition of Narayanganj lake
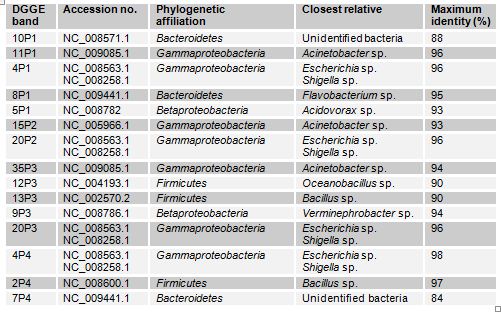
In case of Narayanganj lake four samples were analyzed through DGGE. The bacterial phyla that were found to be present in different sites of this lake are Bacteroidetes, Gammaproteobacteria, Betaproteobacteria and Firmicutes (Table 3.2). The Gammaproteobacteria was the dominant phylum in this site. In these samples prominent bands according to band intensity in the DGGE gel (Figure 3.1) were most closely related to known organisms in the database included Acidovorax sp. (5P1), Acinetobacter sp. (band 11P1), Bacillus sp. (13P3), the Escherichia-Shigella group (4P1), Oceanobacillus sp.(12P3), Verminephrobacter sp. (9P3). One prominent band (band 10P1) was most closely related to unidentified bacteria at identity level of 88%. Percent identities ranged between good (98%) and poor (84%).
Table: Identity of phylotypes in DGGE profiles of freshwater samples in Mirpur lake (Figure)
Community composition of Gulshan lake
The bacterial phyla that were found in Gulshan lake were Bacteroidetes, Gammaproteobacteria, Betaproteobacteria, Actinobacteria and Firmicutes (Table 3.3). The Gammaproteobacteria was the dominant phylum in this site also. In these samples prominent bands according to band intensity in the DGGE gel (Figures 3.1 and 3.2) were most closely related to known organisms in the database included the Escherichia-Shigella group (4G1), Acinetobacter sp. (band 14G1), Aeromonas sp. (16G1) Bacteroides sp. (17G2), Streptococcus sp. (18G2), Polaromonas sp. (19G2), Vibrio sp. (47G3), Staphylococcus sp. (40G4). Two prominent bands (bands 43G3 and 43G4) were most closely related to unidentified bacteria. Percent identities ranged between good (99%) and poor (84%).
Table: Identity of phylotypes in DGGE profiles of freshwater samples in Narayanganj lake (Figure)
Community composition of Ramna lake
In total nine samples were examined using DGGE. The bacterial phyla (Table 3.4) present were Gammaproteobacteria, Bacteroidetes, Deltaproteobacteria and Firmicutes. The dominant phylum was the Gammaproteobacteria in this site. In these samples prominent bands according to band intensity in the DGGE gel (Figures 3.1 and 3.2) were most closely related to known organisms in the database included the Escherichia-Shigella group (band 4R1), Yersinia sp. (21R1), Acinetobacter sp. (15R1), Pelobacter sp. (22R2), Vibrio sp. (42R8), Oceanobacillus sp. (44R9), Staphylococcus sp. (40R5) and Alkalilimnicola sp. (45R11). Four prominent bands (7R1, 49R3, 49R5 and 43R11) were most closely related to either uncultured or unidentified bacteria. Percent identities ranged between good (99%) and poor (84%). Four bands of sample R3, R5, R8 and R11 were most closely related to only Shigella species (96-98% identity) of the E. coli- Shigella group.
Community composition of Dhanmondi lake
Eleven samples collected from different sites of Dhanmondi lake were examined. The bacterial phyla (Table 3.5) those were present: Gammaproteobacteria, Actinobacteria, Cyanobacteria, Betaproteobacteria, Bacteroidetes and Firmicutes. The dominant phylum was the Gammaproteobacteria in this site. In these samples prominent bands according to band intensity in the DGGE gel (Figures 3.1 and 3.2) was most closely related to known organisms in the database included the Escherichia-Shigella group (band 29D1), Oceanobacillus sp. (26D1), Pseudomonas sp. (27D1), Shewanella sp. (28D1), Synechococcus sp. (34D1), Streptococcus sp. (18D1), Yersinia sp. (32D3), Flavobacterium sp. (8D3), Acinetobacter sp. (25D4), Verminephrobacter sp. (9D3), Bacillus sp. (2D5), and Arthrobacter sp. (6D5). Percent identities ranged between good (99%) and poor (82%). Eight prominent bands (sample D1, D3, D7, D9, D10 and D11) were most closely related to unidentified bacteria. Five bands of sample D7, D9, D10, D11 and D12 were most closely related to only Shigella species (98% identity) of the E. coli- Shigella group. Among all the samples of all the sites D1 showed the highest number or bacteria with nine prominent bands representing nine bacteria.
Community composition of Gafargaon lake
Three samples collected from different sites of Gafargaon lake were examined using DGGE. The bacterial phyla (Table 3.6) present were Gammaproteobacteria and Firmicutes. The Gammaproteobacteria was dominant phylum in this site. In these samples prominent bands according to band intensity in the DGGE gel (Figure 3.2) were most closely related to known organisms in the database included the Escherichia-Shigella group (band 36F2), Vibrio sp. (42F3), Bacillus sp. (41F3), Staphylococcus sp. (40F1) and Alkalilimnicola sp. (45F3). Three prominent bands (39F1, 43F2 and 45F3) were most closely related to either uncultured or unidentified bacteria. Percent identities ranged between good (99%) and poor (85%).Another band, 37F3, was most closely related to only Shigella species (98% identity) of the E. coli- Shigella group.
Statistical analysis
NPar Tests
The NPar test was performed to test normal parameters, extreme differences and Kolmogorov-Smirnov Z. The Kolmogorov-Smirnov test compares an observed cumulative distribution function with a theoretical cumulative distribution. The output indicate that the test distribution is normal and uniform.
Analysis of the occurrence of the phyla with respect to the enrichment procedures
A graph (Figure 3.10) was prepared through SPSS to compare the presence of the seven phyla e.g. Gammaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Actinobacteria, Cyanobacteria, Bacteroidetes and Firmicutes in different enrichment media, 1.0× enrichment, 0.1× enrichment (diluted) and the direct sample.
Bivariate correlations between the genes examined by PCR
To find out the presence of any correlation between the specific virulent genes of E. coli and Shigella, the bivariate correlation was done. The correlation Table (Appendix-IV) displays Pearson correlation coefficients, significance values, and the number of cases with non-missing values. The Table shows that correlation is significant between ipaH and ial genes (p = 0.001), ipaH and bfp genes (p = 0.089) and also between ipaBCD and virA genes (p = 0.029). Correlation is significant at 0.1 level.
Bivariate correlations between the seven phyla
The bivariate correlation was done to find out correlation between the seven phyla present in the samples e.g., Gammaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Actinobacteria, Cyanobacteria, Bacteroidetes and Firmicutes. The correlation Table (Appendix-IV) shows that there is no significant correlation between the presence of Gammaproteobacteria and other six phyla.
Bivariate correlations between the bacterial genera
The bivariate correlation was done to find out correlation between the twenty one different bacterial genera. Significant correlation was absent between the E. coli-Shigella group and other bacterial genera identified in the samples.
Occurrence of different phyla with respect to specific virulent genes
There is an important association between the presence of different phyla and the presence of the ipaH, ipaBCD, ial and virA genes.
In the studied samples Gammaproteobacteria was the dominant phylum and the other six phyla found to be present in the samples are Betaproteobacteria, Deltaproteobacteria, Actinobacteria, Cyanobacteria, Bacteroidetes and Firmicutes. The number of Actinobacteria, Deltaproteobacteria and Cyanobacteria was very low. The phyla Firmicutes, Betaproteobacteria and Bacteroidetes were present with Gammaproteobacteria in maximum cases.















