Protein-protein interaction prediction is a field that combines bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein-protein interactions is critical for investigating intracellular signaling pathways, modeling protein complex structures, and gaining insights into various biochemical processes.
Scientists have collaborated to develop a structurally motivated deep learning method based on recent advances in neural language modeling. D-SCRIPT, the team’s deep-learning model, was able to predict protein-protein interactions (PPIs) from primary amino acid sequences.
Understanding protein-protein interactions (PPIs) is critical for understanding cell physiology in both normal and disease states because PPIs are required for almost every process in a cell. It is also important in drug development because drugs can affect PPIs. Protein-protein interaction networks (PPIN) are mathematical representations of physical interactions between proteins in a cell.
Professor Lenore Cowen of the Tufts Department of Computer Science and colleagues from the Massachusetts Institute of Technology (MIT) collaborated to design a structurally-motivated deep learning method based on recent advances in neural language modeling in a study published in the journal Cell Systems. D-SCRIPT, the team’s deep-learning model, predicted protein-protein interactions (PPIs) from primary amino acid sequences.
These predictions enable researchers to use a clustering method to model PPI networks and detect functional subnetworks, or modules. Scientists study PPI networks in organisms to better understand their signaling circuitry, which could lead to better predictions of cell behavior and gene functions while discovering functional modules in PPI networks could help researchers gain a better understanding of the cellular functional organization.
Cowen, along with MIT’s Computer Science and Artificial Intelligence Lab researchers Sam Sledzieski, Rohit Singh, and renowned computational biologist Bonnie Berger, discovered that the D-SCRIPT model, trained on more than 38,000 human PPIs, was more generalizable than the current state-of-the-art approach (the deep-learning method PIPR), and thus could characterize fly proteins. They also used D-SCRIPT to look for PPIs related to cow digestion and found functional gene modules related to immune response and metabolism.
The interactome is the collection of PPIs that occur in a cell, organism, or biological context. The advancement of large-scale PPI screening techniques, particularly high-throughput affinity purification combined with mass spectrometry and the yeast two-hybrid assay, has resulted in an explosion of PPI data and the construction of increasingly complex and complete interactomes. The availability of PPI prediction algorithms supplements this experimental evidence. Many of these details can be found in molecular interaction databases like IntAct.
The researchers concluded that the D-SCRIPT model, which was trained on human PPI data, could be applied to a wide range of species of interest, including those that have been little studied or lack PPI data.
Protein-protein interactions (PPIs) are the foundation of many important cellular processes, including signal transduction, molecular transport, and various metabolism pathways, whereas abnormal PPIs are the foundation of multiple aggregation-related diseases, including Alzheimer’s disease, and may lead to cancer. As a result, PPIs have been extensively studied in the fields of bioscience and medical research.
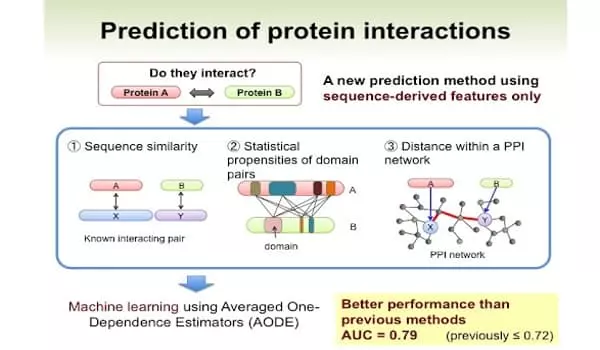
The field of protein-protein interaction prediction is closely related to the field of protein-protein docking, which attempts to fit two proteins with known structures into a bound complex using geometric and steric considerations. This is a useful model of investigation when both proteins in the pair have known structures and are known (or strongly suspected) to interact; however, because so many proteins lack experimentally determined structures, sequence-based interaction prediction methods are especially useful in conjunction with experimental studies of an organism’s interactome.