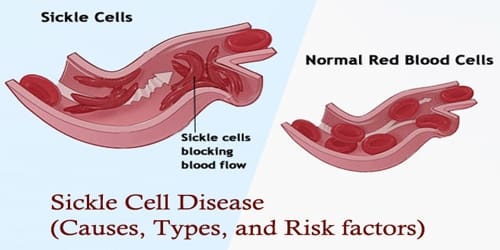
Sickle Cell Disease (Causes, Types, and Risk factors)
Definition: Sickle cell disease (SCD) or Sickle cell anemia is a group of disorders that affects hemoglobin, the molecule in red blood cells (RBCs) that delivers oxygen to cells throughout the body. Normally, RBCs are shaped like discs, which give them the flexibility to travel through even the smallest blood vessels.
Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain (“sickle cell crisis”), anemia, swelling in the hands and feet, bacterial infections and stroke. Long-term pain may develop as people get older. The average life expectancy in the developed world is 40 to 60 years.
Signs and symptoms of sickle cell disease usually begin in early childhood. Characteristic features of this disorder include a low number of red blood cells (anemia), repeated infections, and periodic episodes of pain. The severity of symptoms varies from person to person. Some people have mild symptoms, while others are frequently hospitalized for more serious complications.
There’s no cure for most people with sickle cell anemia. But treatments can relieve pain and help prevent problems associated with the disease.
About 90% of people survive to age 20, and close to 50% survive beyond age 50. In 2001, according to one study performed in Jamaica, the estimated mean survival for people with the sickle cell was 53 years old for men and 58 years old for women with homozygous SCD. The specific life expectancy in much of the developing world is unknown.
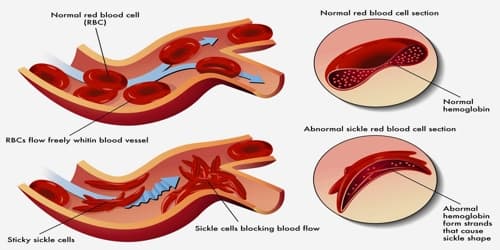
Causes and Types of Sickle cell disease (SCD): Sickle cell anemia is caused by a mutation in the gene that tells our body to make the red, iron-rich compound that gives blood its red color (hemoglobin). Hemoglobin allows red blood cells to carry oxygen from our lungs to all parts of our body. In sickle cell anemia, the abnormal hemoglobin causes red blood cells to become rigid, sticky and misshapen.
The four main types of sickle cell anemia are caused by different mutations in these genes.
- Hemoglobin SS disease – Hemoglobin SS disease is the most common type of sickle cell disease.
- Hemoglobin SC disease – Hemoglobin SC disease is the second most common type of sickle cell disease.
- Hemoglobin SB+ (beta) thalassemia – Hemoglobin SB+ (beta) thalassemia affects beta-globin gene production.
- Hemoglobin SB 0 (Beta-zero) thalassemia – Sickle beta-zero thalassemia is the fourth type of sickle cell disease. It also involves the beta-globin gene.
Hemoglobin SD, hemoglobin SE, and hemoglobin SO – These types of sickle cell disease are rarer and usually don’t have severe symptoms.
In people with sickle cell disease, at least one of the beta-globin subunits in hemoglobin is replaced with hemoglobin S. In sickle cell anemia, which is a common form of sickle cell disease, hemoglobin S replaces both beta-globin subunits in hemoglobin. In other types of sickle cell disease, just one beta-globin subunit in hemoglobin is replaced with hemoglobin S. The other beta-globin subunit is replaced with a different abnormal variant, such as hemoglobin C.
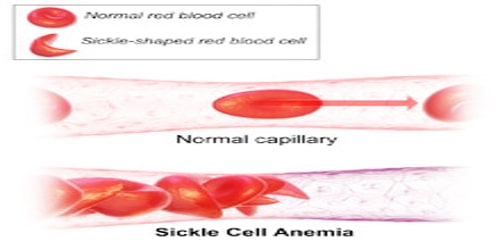
Abnormal versions of beta-globin can distort red blood cells into a sickle shape. The sickle-shaped red blood cells die prematurely, which can lead to anemia. Sometimes the inflexible, sickle-shaped cells get stuck in small blood vessels and can cause serious medical complications.
Risk factors and Complications: As of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait. About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa. It also occurs relatively frequently in parts of India, the Arabian Peninsula and among people of African origin living in other parts of the world.
Children are only at risk for sickle cell disease if both parents carry sickle cell trait. A blood test called a hemoglobin electrophoresis can also determine which type people might carry. People from regions that have endemic malaria are more likely to be carriers.
Sickle cell disease (or Sickle cell anemia) can lead to a host of complications, including:
- Stroke
- Pulmonary hypertension
- Acute chest syndrome
- Organ damage
- Blindness
- Leg ulcers
- Gallstones
- Priapism
Anemia is a shortage of RBCs. Sickle cells are easily broken. This breaking apart of RBCs is called chronic hemolysis. RBCs generally live for about 120 days. Sickle cells live for a maximum of 10 to 20 days.
Information Source: